LES PRODROGUES,
OU PROMEDICAMENTS *
Gérard GOMEZ
Plan de
l'étude :
1) Définition
3) Devenir, dans l'organisme, d'un médicament administré par voie
orale
3-1) Généralités
3-2) Dissolution
du médicament
3-3) Franchissement
de la barrière gastro-intestinale
3-4) La
suite du trajet du médicament
4-1) La
pivampicilline
4-2) Le
valaciclovir
4-3) L'azathioprine
4-4) La
L-Dopa
5) Conclusion
1) Définition :
Le mot
drogue est pris ici dans le sens du mot anglais drug c'est-à-dire
médicament.
Une
prodrogue est une molécule constituant le principe actif d'un médicament, liée
chimiquement (par liaison covalente) à un groupe d'atomes.
Cette prodrogue
est pharmacodynamiquement inactive ou beaucoup moins active que le principe
actif qu'elle contient.
Dans
l'organisme une biotransformation libère le principe actif et lui permet de
jouer son rôle thérapeutique ; le groupe d'atomes libéré concomitamment ne doit
bien sûr avoir aucune action pharmacologique ni aucune toxicité.
Un
exemple
:
Le
propacétamol
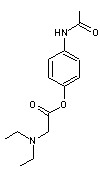
est une
prodrogue du paracétamol (à usage intraveineux) qui est hydrolysée dans
l'organisme par les estérases plasmatiques (enzymes du plasma qui opèrent
l'hydrolyse des esters) en
- paracétamol
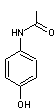
et en
- N,N-diéthylglycine qui est un
acide aminé
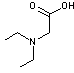
On
élabore des prodrogues pour plusieurs raisons que l'on va énumérer ; on peut
rechercher l'un de ces effets à l'exclusion des autres ou plusieurs d'entr'eux
simultanément :
- On aide le principe actif à
franchir les barrières de l'organisme (intestinales, hématoencéphaliques,
cutanées).
- On modifie la vitesse du
métabolisme du médicament (effet retard ou effet prolongé) ;
- On protège certaines molécules de
l'acidité prononcée de l'estomac ou de l'activité de certaines enzymes qui les
détruiraient.
Remarque : cet
effet peut aussi être obtenu autrement, grâce à certaines formulations
galéniques.
- On souhaite modifier les
propriétés organoleptiques ; on masque l'odeur ou le goût (amertume ….) du
médicament.
- On souhaite par exemple que le
médicament soit solide plutôt que liquide pour faciliter son conditionnement.
- ….
Essayons
à titre d'exemple de comprendre comment une prodrogue peut aider le principe
actif à franchir une barrière de l'organisme, par exemple la barrière
gastro-intestinale.
Pour
cela il faut examiner le devenir d'un médicament administré par voie orale.
3) Devenir, dans l'organisme, d'un médicament administré par voie
orale :
3-1) Généralités
:
Lorsqu'un
médicament est pris par voie orale, il doit passer dans le sang pour agir ; il
peut le faire à plusieurs niveaux :
- au niveau buccal car la face
inférieure de la langue est tapissée par une muqueuse très mince et fortement
vascularisée. La surface d'absorption est faible ; le pH se situe entre 6,7 et
7,0.
- au niveau de l'estomac, la muqueuse
est très épaisse et la vascularisation veineuse faible ; le pH se situe entre 2
et 3 ; la surface d'absorption est faible (environ 0,11 m2).
- au niveau de l'intestin qui a une
surface d'absorption très importante (environ 300 m2 ) et une très forte
vascularisation le pH est entre 7 et 8.
En
tenant compte de ces données on voit que les médicaments passent dans le sang,
en très grande majorité, au niveau de la voie gastro-intestinale, surtout à partir
de l'intestin, assez peu au niveau de l'estomac.
Le
schéma général du trajet des nutriments et des médicaments est donc le suivant
:
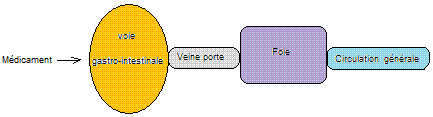
Dans un
premier temps il est nécessaire qu'un médicament se dissolve dans le milieu
gastro-intestinal ; dans un second temps qu'il diffuse à travers la paroi
gastro-intestinale pour retrouver après un premier passage dans le foie, la
circulation sanguine systémique.
On va
examiner tour à tour ces deux phases, la dissolution puis la diffusion à travers la paroi.
3-2) Dissolution
du médicament :
C'est
la solubilité en milieu aqueux (hydrosolubilité) qui va être importante dans
cette première phase.
Exemples :
- l'acétylsalicylate de lysine (Aspégic
®)
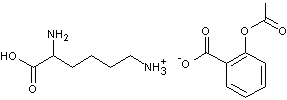
est un
composé plus ionisé en milieu aqueux que l'acide acétylsalicylique (Aspirine
®)
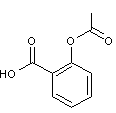
On trouve,
en effet dans le sel de lysine des groupes –COO- et des groupes NH3+.Cela
le rend plus soluble en milieu aqueux gastro-intestinal et globalement la
résorption est plus rapide et plus complète ; des taux sanguins efficaces sont plus rapidement atteints.
- Le propacétamol cité plus haut qui
est un ester du paracétamol (réaction de la fonction acide carboxylique de
l'acide aminé diéthylglycine avec la fonction phénol du paracétamol) est plus
soluble dans l'eau que le paracétamol seul.
La
formulation galénique peut parfois permettre de contrôler plusieurs facteurs
concernant cette mise en solution :
- A quel niveau elle
aura lieu ; on peut, par exemple, pour éviter que le médicament ne se dissolve
dans l'estomac (pH 1 à 3,5) et donc le protéger d'une trop grande acidité
(principe actif susceptible d'être dégradé par un pH trop faible ou mauvaise
résorption au niveau gastrique) l'enrober d'une substance qui lui permet
d'atteindre l'intestin (pH 7 à 8) sans être détruit (forme
gastro-résistante).L'enrobage gastro-résistant et entéro-soluble peut être par
exemple l'acéto-phtalate de cellulose (annexe1).
- On peut aussi si on
souhaite une libération prolongée (pour éviter à l'organisme des pics de
concentration) enfermer le principe actif dans une trame qui lui permettra de
n'être libéré que petit à petit (annexe 2).
3-3) Franchissement
de la barrière gastro-intestinale :
Après
leur mise en solution, les médicaments doivent franchir la paroi gastro-intestinale
pour se retrouver dans le sang.
Il
s'agit de franchir un ensemble de cellules qui forment cette paroi.
Une
cellule humaine est délimitée par une membrane plasmique (qui sépare le
cytoplasme du milieu extérieur) ainsi structurée :
- Une bicouche phospholipidique
faite d'un ensemble de molécules de phosphatidylcholine (voir figure
ci-dessous) dont les chaînes carbonées sont à l'intérieur et constituent une
phase hydrophobe continue et les têtes hydrophiles placées à l'extérieur.
La bicouche
possède une certaine fluidité liée à la température, mais aussi à la nature des
acides gras qui la constituent ; ceux insaturés à chaîne courte augmentent
cette fluidité.
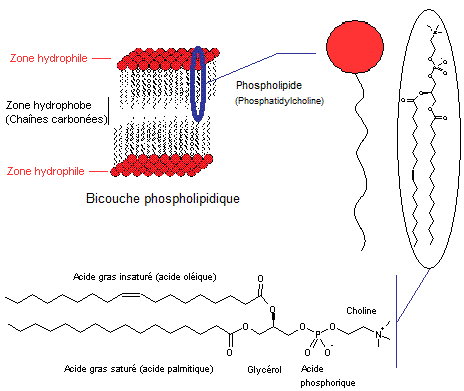
- Des protéines dites membranaires
sont enfoncées dans la bicouche, ou traversent celle-ci (elles sont alors dites
transmembranaires) ; on trouve aussi des protéines simplement placées autour de
la bicouche qu'on appelle protéines périphériques.
v
Ces protéines servent de récepteurs.
v
Elles assurent les échanges d'ions (Na+,
K+, …) entre la cellule et son environnement.

Cet ensemble,
qui globalement est composé de 60% de phospholipides et 40% de protéines,
forme une membrane pratiquement
imperméable.
Les
médicaments devront donc être résorbés et ceci pourra se faire de différentes
façons :
Les
molécules traversent la paroi spontanément dans le sens du gradient (du milieu
le plus concentré vers le milieu le moins concentré).
Ce mode
de diffusion est régi par la loi de Fick :
![]()
où :
- V est le nombre de molécules
traversant la paroi par unité de temps (flux)
- D le coefficient de diffusion de
la molécule à travers la paroi
- S la surface de la paroi
- Ce la concentration de la molécule
à l'intérieur du tube digestif
- Ci la concentration de la molécule
dans le sang
- a l'épaisseur de la paroi.
Cette
formule montre que le flux de diffusion est d'autant plus grand que :
- S est grande (surface d'échanges)
- (Ce-Ci) est grande (différence de concentration
en molécules qui diffusent entre l'extérieur du vaisseau sanguin et l'intérieur
de celui-ci)
- a l'épaisseur de la paroi du
vaisseau est plus petite
- D le coefficient de diffusion est
grand.
On montre que D est d'autant plus grand que la
molécule est soluble dans la paroi ; ce qui implique compte-tenu de la nature
de la bicouche que la molécule ne soit pas ionisée ni même trop polaire,
qu'elle soit liposoluble donc lipophile, avec un poids moléculaire faible ou
moyen (inférieur à 500 Daltons).
Exemple : Le glucose et les
acides aminés ne peuvent diffuser par diffusion passive à cause des polarités
de leurs molécules ; le glucose (et les glucides en général) à cause des
groupes –OH et les acides aminés à cause de leur fonction acide carboxylique
–COOH.
Remarques :
Ø
La lipophilie d'une molécule est son
aptitude à se dissoudre dans une matière grasse. La présence de groupes méthyle
(-CH3), éthyle (-C2H5), méthyne =CH- ou les
noyaux aromatiques confèrent à une molécule un caractère lipophile. Au
contraire les groupements –OH, -COOH, C=O, et les groupes ionisés comme COO-
ou NH3+ confèrent à la molécule un caractère hydrophile
donc lipophobe. On apprécie la lipophilie d'une molécule ou plutôt sa balance
lipophile/hydrophile en exprimant le coefficient de partage P de cette molécule
dans un mélange octan-1-ol/eau c'est-à-dire le rapport entre respectivement les
concentrations dans l'octan-1-ol et l'eau. Plus log(P) est élevé et plus la
molécule est lipophile.
Pour avoir quelque
chance de traverser une membrane plasmique par diffusion passive, log(P) doit
être compris entre 1 et 5. La valeur optimale pour une absorption orale étant
1,8.
La noradrénaline joue
le rôle d'hormone adrénergique et de neurotransmetteur.
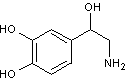
Elle a un log(P) = -2
(P = 0,01). Elle est donc 100 fois plus soluble dans l'eau que dans un solvant
organique. Elle est très hydrophile et passe mal la membrane cellulaire.
La chlorpromazine est
un psychotrope utilisé dans le traitement des schizophrénies et des phases du
trouble bipolaire
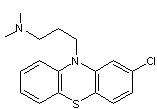
Elle doit passer la
barrière hémato-encéphalique riche en graisse ; elle a un log(P) =5. Elle est très
liposoluble.
Ø
L'ionisation d'une molécule qui est un
facteur défavorable à la traversée d'une membrane, est bien sûr liée au pH du
milieu environnant. Ainsi prenons l'aspirine dont le pKa de la fonction acide
carboxylique est 3,5 ; au niveau gastrique de pH = 1 elle est totalement non
ionisée et donc bien résorbée, tandis qu'au niveau du duodénum (pH = 7,4) elle
est totalement ionisée et la diffusion passive est beaucoup plus difficile.
Certaines
molécules ne peuvent passer les membranes (gastro-intestinale ou
hémato-encéphalique) car ionisées ou peu lipophiles. Une protéine de transport
membranaire va sélectivement reconnaître cette molécule comme étant proche de
son substrat endogène et l'aider à
traverser la membrane.
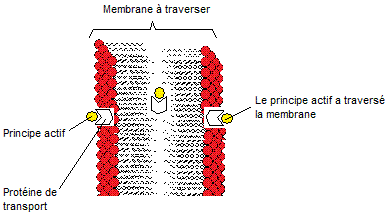
Dans
certains cas, le transport se fait contre le gradient de concentration et
l'énergie nécessaire pour ce faire est fournie par l'hydrolyse de l'ATP :
ATP →
ADP + Pi (Phosphate inorganique) + 8 kcal.mol-1.
3-4) La suite
du trajet du médicament :
Parvenu
dans le sang un médicament va être distribué dans l'organisme par la
circulation générale et il atteint sa cible où il peut alors agir.
Lorsqu'il
s'agit d'une prodrogue, la libération du principe actif intervient parfois sous
l'effet des enzymes de la muqueuse intestinale, ou parfois au niveau du foie.
La
biodisponibilité d'un médicament est la fraction de dose administrée ou du
principe actif libéré qui parvient sous forme inchangée dans la circulation
sanguine systémique.
Cette
biodisponibilité dépend de beaucoup de facteurs ; pour l'aspirine par exemple
elle dépend de la dose absorbée ; elle est de 60% pour moins de 500 mg et 90%
pour 1g ou plus. On l'explique par le fait que cette molécule est hydrolysée au
niveau hépatique en acide salicylique ; à forte dose, il y a saturation de
l'hydrolyse hépatique ce qui permet à plus de molécules non transformées de
continuer dans la voie sanguine.
4) Retour sur les prodrogues :
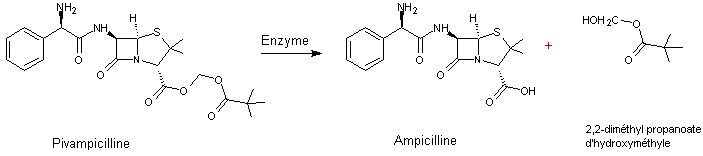
4-1) La
pivampicilline
est une
prodrogue ; l'ampicilline en est le principe actif.
L'ampicilline
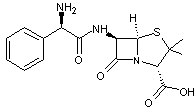
est un
antibiotique, une aminopénicilline à spectre large ; elle couvre tout le
spectre des pénicillines conventionnelles (comme l’amoxicilline) plus beaucoup
d’autres souches comme les entérocoques.
Par
absorption orale, il n'y a que 40% de la dose administrée qui est absorbée par
l'organisme. La raison de cette mauvaise résorption réside dans la présence du
groupe acide carboxylique –COOH qui diminue sa lipophilie.
Afin
d'améliorer l'absorption de ce principe actif, on le transforme en
pivampicilline
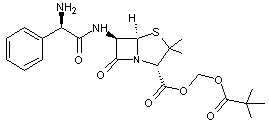
et son
taux d'adsorption devient 98%.
On a
transformé la fonction acide carboxylique de l'ampicilline en ester ce qui a
fait disparaître l'ionisation due à la fonction acide. La molécule de pivampicilline
est beaucoup plus lipophile et traverse facilement la bicouche lipidique.
Une
fois dans le sang la prodrogue se transforme totalement en ampicilline par
hydrolyse enzymatique :
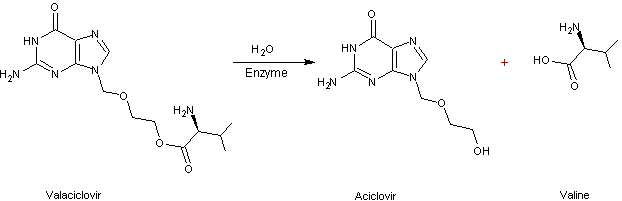
4-2) Le
valaciclovir
est une
prodrogue dont l'aciclovir est le principe actif.
L'aciclovir
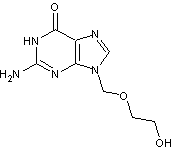
est un
antivirus qui permet de bloquer la multiplication du virus de l'herpès dans les
cellules infectées. Il est n'est que partiellement résorbé dans le tractus
intestinal, la biodisponibilité est comprise entre 15 et 30%.
Le
valaciclovir
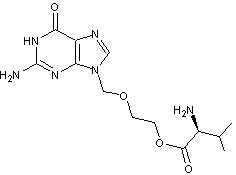
lui, est
bien résorbé et est rapidement et presque intégralement transformé en aciclovir
et valine ; la biodisponibilité de l'aciclovir issu du valaciclovir (1000mg)
est de 54%.
C'est
la fonction alcool (-OH) de l'aciclovir qui est la cause de la faible lipophilie
du principe actif ; en l'estérifiant par la fonction acide carboxylique de la
valine, on fait disparaître la polarité et la lipophilie augmente.
Dans le
sang l'hydrolyse de la prodrogue, restitue l'aciclovir et la valine.
4-3) L'azathioprine
C'est
la prodrogue de la 6-mercaptopurine.
La
6-mercaptopurine
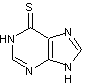
possède
des propriétés immunosuppressives et cytostatiques que l'on utilise dans le
traitement des leucémies.
Sa
biodisponibilité est faible, en moyenne de 16% (de 5 à 37%).
L'azathioprine
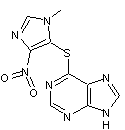
est
très rapidement et presque entièrement résorbé dans le système digestif.
Sa
transformation rapide en 6-mercaptopurine est assurée par une ou plusieurs
Glutathion-S-Transférase (GST) :

4-4) La
L-Dopa
La
L-Dopa est la prodrogue de la dopamine.
La
dopamine
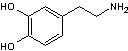
est un
neurotransmetteur (messager chimique entre les neurones) jouant un rôle
important dans le fonctionnement du cerveau. Le manque de dopamine dans le
système nerveux central provoque la maladie de Parkinson.
La maladie de Parkinson touche une zone du cerveau appelée "locus
niger" (substance noire). Cette affection neurodégénérative se caractérise
par la destruction progressive des neurones produisant la dopamine impliquée
dans le contrôle des mouvements. Il en résulte une diminution significative de
celle-ci, diminution responsable des symptômes (notamment moteurs) de la
maladie.
La
dopamine que l'on pourrait apporter par voie orale ne pourrait passer la
membrane hémato-encéphalique à cause de sa lipophilie médiocre.
On
administre donc de la lévodopa (L-dopa)
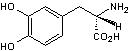
qui
peut traverser la membrane hémato-encéphalique par transport actif, grâce au
transporteur LAT-1 (L-Amino-acid Transporter 1) qui est une protéine permettant
le transport au travers des membranes des acides aminés neutres à longue
chaîne.
Parvenue
dans le système nerveux, la prodrogue est libérée de son transporteur et
transformée en dopamine par décarboxylation enzymatique.
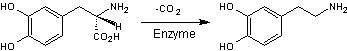
La
dopamine ainsi formée permet d'atténuer les symptômes de la maladie de
Parkinson.
Remarques :
- La L-Dopa traverse la membrane intestinale
par un mécanisme analogue, en utilisant un transporteur commun à plusieurs
acides aminés.
- 50 à 70% de la L-Dopa absorbée est
décarboxylée au niveau intestinal par la Dopa décarboxylase. Une partie est
aussi décarboxylée dans le cerveau avant d'atteindre les noyaux gris centraux.
Souvent on associe lors de la prise de la L-Dopa, celle d'un inhibiteur de la
Dopa décarboxylase qui empêche la transformation de la L-Dopa avant qu'elle
n'atteigne sa cible, ce qui diminue beaucoup la dose administrée pour obtenir
le même effet clinique.
5) Conclusion :
Les
prodrogues permettent, dans beaucoup de cas, d'augmenter l'efficacité de
principes actifs déjà utilisés ; elles s'avèrent donc très utiles à un moment
où il devient de plus en plus difficile de trouver des principes actifs
nouveaux.
Annexe 1
Acétophtalate
de cellulose
C'est un
polymère de cellulose constituant, dans certains médicaments, un enrobage qui
protège le principe actif et lui permet d'atteindre l'intestin sans avoir été
dégradé dans l'estomac (enrobage gastro-résistant).
C'est un
dérivé de la cellulose où environ la moitié des groupes –OH de celle-ci sont
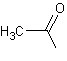
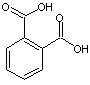
estérifiés par des groupes acétyles, un quart
sont estérifiés par un ou deux carboxyles de l'acide phtalique et le
reste des –OH est inchangé.
Cellulose :

Groupe
acétyle :
Acide
phtalique :
Structure
d'un comprimé permettant la libération prolongée d'un médicament (système OROS
® push-pull)
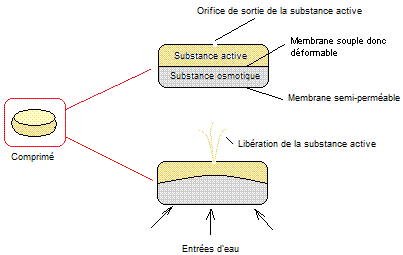
Le principe
d'action est le suivant :
Plongé dans
un liquide moins concentré que la substance osmotique la membrane
semi-perméable qui constitue l'enveloppe du comprimé se laisse traverser par
celui-ci ; il augmente le volume du compartiment inférieur et la surface de
séparation des deux compartiments en se déformant pousse la substance active à
l'extérieur par l'orifice de sortie. Cette sortie s'effectue à vitesse
constante et est indépendante du pH du liquide qui traverse la membrane
semi-perméable. Le délai d'action (temps qu'il faut pour que l'expulsion de la
substance active débute) est d'environ 2 heures ; la durée de libération de la
substance active est environ 8 heures.
- La membrane semi-perméable est
constituée d'acétate de cellulose :
La cellulose
est un polymère de formule générale
Un ou
plusieurs groupes –OH sont estérifiés par l'acide éthanoïque (CH3COOH).
- La substance osmotique est
constituée de chlorure de sodium (NaCl) et d'un dérivé cellulosique.
- L'orifice de sortie de la
substance active a un diamètre qui varie entre 250 et 500µm.