MECANISMES REACTIONNELS
1) Définition
2) Réactions
homolytiques - Réactions hétérolytiques
3) Réactifs nucléophiles - Réactifs
électrophiles
4) Les intermédiaires
réactionnels
5) Quelques
mécanismes de réactions d'addition
6) Quelques mécanismes de réactions de
substitution
7) Les
réactions d'élimination
1) Définition
:
C'est une suite chronologique d'étapes élémentaires
qui conduisent des réactifs aux produits, contrairement à
l'équation bilan qui affiche seulement, les réactifs d'un côté, les produits de
l'autre.
Exemple:
Polymérisation de l'éthylène:
- bilan :
![]()
- mécanisme :
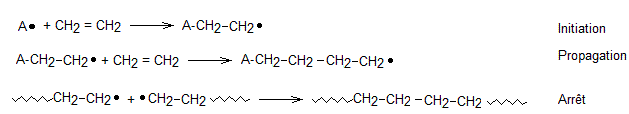
2) Réactions homolytiques -
Réactions hétérolytiques :
réaction
homolytique (ou radicalaire) :
![]() rupture
symétrique de la liaison covalente (formation de radicaux). Cette rupture est
en général initiée par des radiations U.V.
rupture
symétrique de la liaison covalente (formation de radicaux). Cette rupture est
en général initiée par des radiations U.V.
réaction
hétérolytique :
rupture dissymétrique de la liaison covalente
![]()
3) Réactifs nucléophiles - Réactifs électrophiles (dans
les réactions hétérolytiques) :
3-1) Réactif et substrat :
Deux entités
qui existent au début de la réaction et qui réagissent l'une sur l'autre sont,
si l'on s'en tient à l'équation bilan, deux réactifs.
D'un point de vue "mécanisme", on
distinguera, l'entité principale, celle dont on suivra l'évolution, le devenir
et que l'on qualifiera de substrat et l'entité qui agira sur le
substrat et que l'on appellera réactif.
C'est par rapport à la nature du réactif
(nucléophile ou électrophile) que l'on qualifiera la réaction de nucléophile ou
électrophile.
3-2) Réactifs nucléophiles:
Ils pourront être :
- des molécules :
- des ions :
OH- , RO- , CN-
On les désignera alors par B-
- des ions:
NO2+ , Ar-N2+
,….
On les désignera alors par A+
- des molécules polarisées ( ou des molécules polarisables) :
![]()
![]()
4) Les
intermédiaires réactionnels :
4-1) Les radicaux :
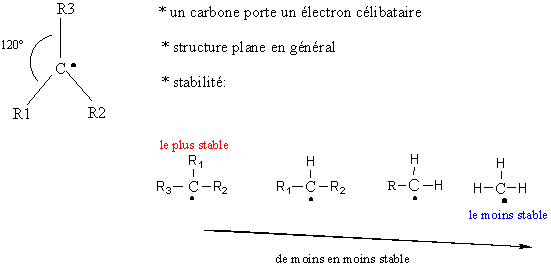
![]()
4-3) Les carbanions :
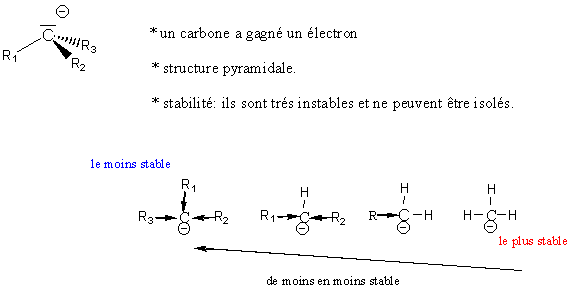
5) Quelques mécanismes de
réactions d'addition:
A) sur les alcènes:
5-1) Addition électrophile :
- bilan :
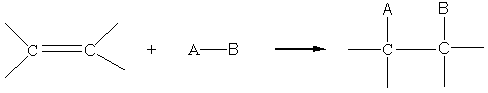
- mécanisme :
o 1ère étape :
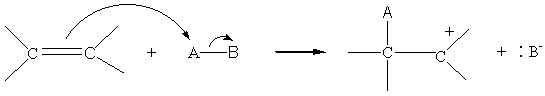
o 2ème étape :
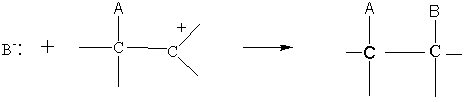
Exemple 1 :
A-B est H-X (halogénure d'hydrogène) ; la polarisation de la molécule![]() du fait de
l'électronégativité de X, nous montre quelle est la partie de la molécule qui
se montre électrophile (H).
du fait de
l'électronégativité de X, nous montre quelle est la partie de la molécule qui
se montre électrophile (H).
Si l'environnement de la double liaison n'est pas
symétrique, il y a deux possibilités d'addition:
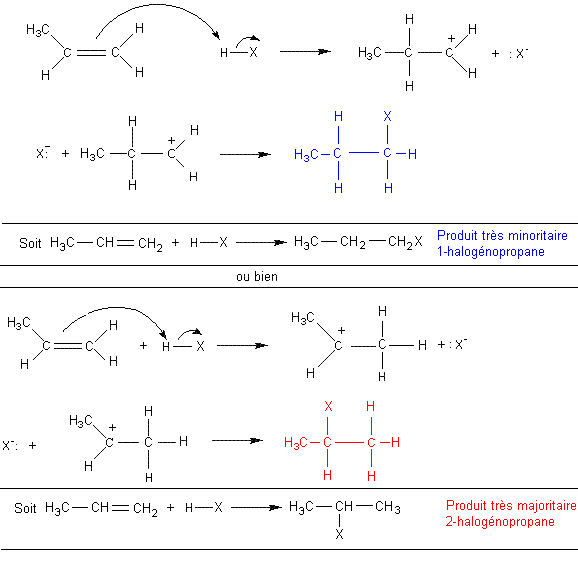
Cette addition est régiosélective; elle se fait
préférentiellement avec fixation de H sur le carbone le moins substitué (règle
empirique de Markovnikov). Cela s'explique par l'effet inducteur répulsif
du groupement CH3- qui "déplace" le doublet de la liaison double
vers le carbone le moins substitué, le rendant plutôt négatif et facilitant de
ce fait l'attaque de l'extrémité électrophile de la molécule de HX.
Exemple 2 :
A-B est X-X (un halogène) : La polarisabilité de la molécule à l'approche d'un
site riche en électrons (double liaison) explique l'apparition d'une extrémité
électrophile de la molécule d'halogène.
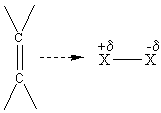
On obtient une anti-addition (on dit aussi souvent
trans-addition)
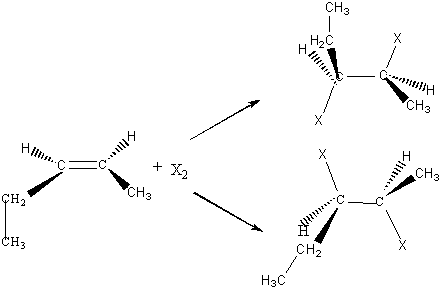
Chaque X s'est fixé de part et d'autre du plan
initial (double liaison).
5-2) Addition
radicalaire: c'est une réaction en chaîne, avec les 3 phases classiques:
- initiation
:
- propagation :
la réaction peut ainsi se
poursuivre avec le radical ainsi régénéré.
- fin : elle peut avoir lieu de plusieurs façons:
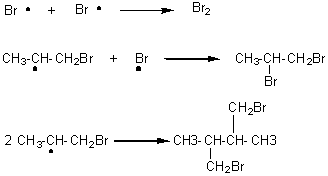
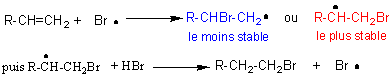
Remarque : contrairement à l'addition électrophile de H-X sur la
double liaison, qui conduisait au composé majoritaire CH3-CHX-CH3,
le composé majoritaire par cette addition radicalaire est CH3-CH2-CH2Br.
Cette addition est anti-Markovnikov (c'est l'effet Kharasch).
B) sur le C=O des composés carbonylés :
5-3) Addition nucléophile :
L' électronégativité de O étant supérieure à celle de
C, la liaison C=O est polarisée C+d = O-d
.
Un réactif B- (nucléophile) va attaquer le
carbone (attaque nucléophile)
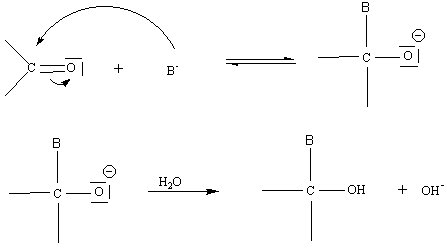
Remarque 1
:en milieu acide, la protonation de l'oxygène, va rendre le carbone plus
positif et favoriser l'attaque par le réactif nucléophile:
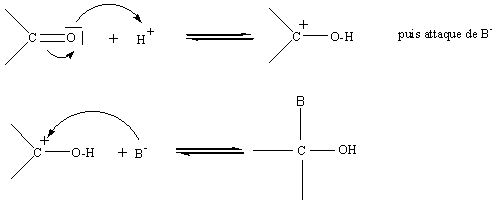
Remarque 2 :
C'est l'étape "attaque par le nucléophile" qui sera toujours la plus
lente des deux.
Remarque 3 :
Souvent, avant l' action de l'eau, il y a fixation du cation A+ du
réactif A-B (qui a donné le nucléophile B-) pour donner
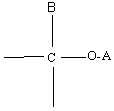
Exemple d'addition
nucléophile sur un carbonyle :
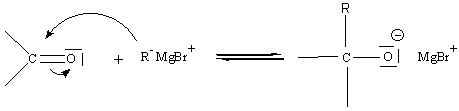
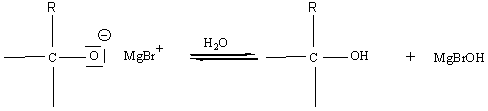
6) Quelques mécanismes de
réactions de substitution :
6-1) sur les halogénoalcanes :
Les dérivés halogénés des alcanes se prêtent bien à
ce genre de réaction (attaque par un réactif nucléophile), du fait de la
polarisation de la liaison R-X due à la grande électronégativité de l'halogène:
![]()
Le bilan d'une telle réaction est
![]() :
:
ou
![]()
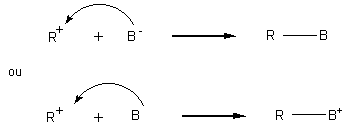
** REACTION SN1
(Substitution Nucléophile monomoléculaire) :
1ère étape : c'est elle qui limitera la vitesse globale de la
réaction :
![]()
Cette dissociation a généralement lieu sous
l'effet d' un solvant polaire ;
puis
2ème étape :
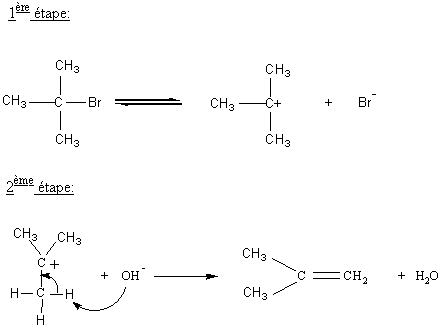
exemple :
action d'une base sur le 2-chloro-2-méthylpropane
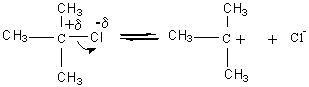
puis
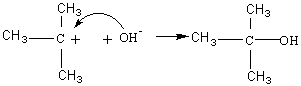
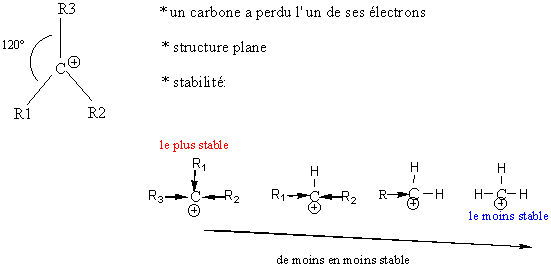
Le carbocation formé à la première étape est d'autant
plus stable que le carbone correspondant est substitué.
Vitesse de réaction :
La vitesse de substitution selon ce mécanisme SN1
augmente donc dans le sens:
R3CX > R2CHX > RCH2X
> CH3X
Le plus
rapide
Le moins rapide
Le facteur cinétique limitant étant la première
étape, on a une loi des vitesses (pour les réactions SN1) de la forme:
v = k [ R-X ] réaction d'ordre 1
Aspect stéréochimique
Le carbocation formé étant plan, l'attaque du réactif
nucléophile peut avoir lieu des 2 côtés du plan; si le carbone est asymétrique,
on aura formation des 2 énantiomères en quantité égale; on aura donc le
racémique.
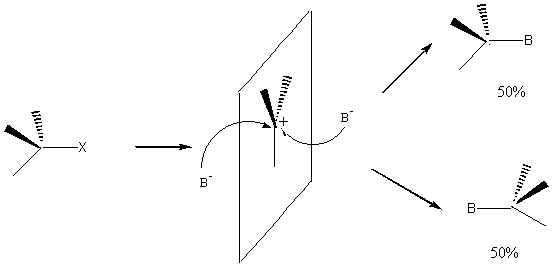
** REACTION SN2 (substitution
nucléophile bimoléculaire) :
une seule étape : 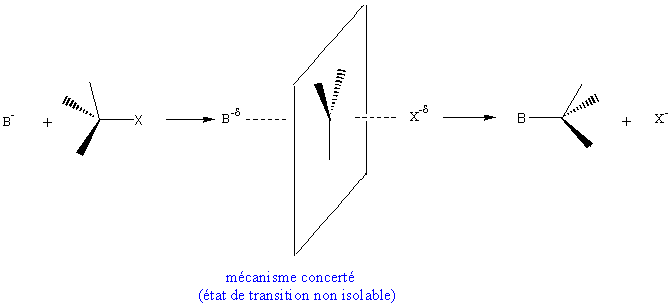
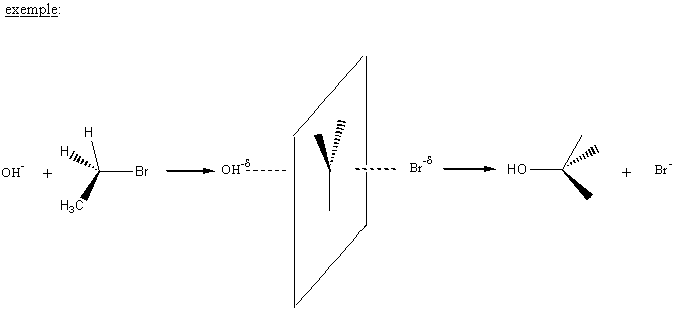
vitesse de réaction :
la vitesse est d'autant plus grande que le composé
est moins substitué:
CH3X > RCH2X >R2CHX
> R3CX
Le plus
rapide
Le moins rapide
v = k [ R-X ] [B-] réaction
d'ordre global 2
Aspect stéréochimique
Si le carbone est asymétrique, la réaction conduit à
une inversion de la configuration ( inversion de WALDEN).
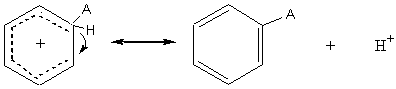
6-2) sur les composés aromatiques :
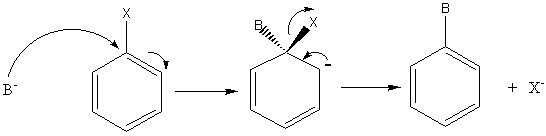
Remarque :
Ces réactions sont difficiles ( la température élevée pour les réaliser, ici
200°C, le montre) car le noyau aromatique est un site riche en e- .
Elles seront plus faciles s'il y a, un, ou des groupements attracteurs d' e-
(-NO2 par exemple) en ortho ou para.
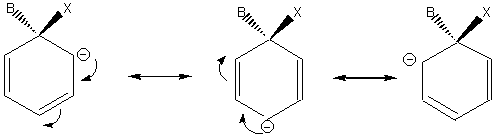
Les différentes formes mésomères de l'ion
intermédiaire formé, contribuent à la stabilité de l'espèce intermédiaire. Plus
il y a de formes mésomères possibles plus la réaction sera facile.
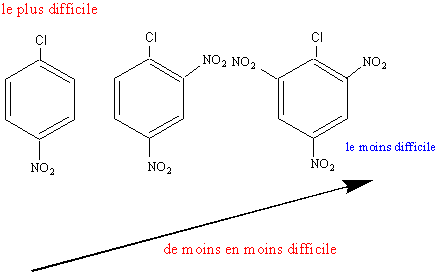
Avec -NO2 en ortho ou para ces
formes sont nombreuses.
Avec -NO2en méta ces formes sont
moins nombreuses.
B) Substitutions électrophiles : fréquentes
sur les composés aromatiques.
- Bilan :
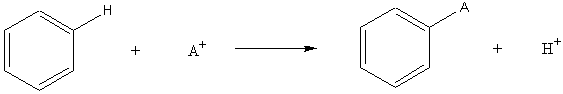
- Mécanisme :
1ère étape : la plus lente
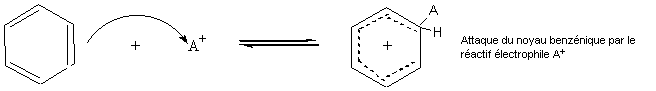
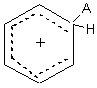
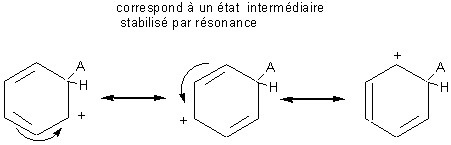
Cet intermédiaire appelé intermédiaire de Wheland
est aussi nommé parfois improprement complexe s.
Remarque : La formation de cet intermédiaire de Wheland est
précédée par la formation d'un complexe P entre
le réactif électrophile et le noyau aromatique :
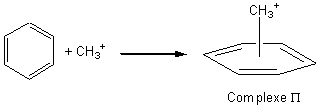
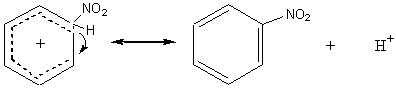
2ème étape: la plus rapide
Exemple :
Nitration du benzène
A+ est NO2+ (ion
nitronium) présent dans l'acide nitrique fumant HNO3 ou formé par
action de H2SO4 concentré sur HNO3 concentré.
1ère étape :
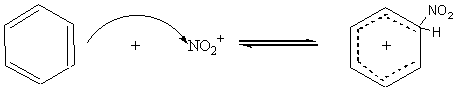
2ème étape :
7) Les
réactions d'élimination :
7-1) Bilan
:Les dérivés halogénés donnent assez souvent aussi des réactions d'élimination;
on les prendra comme exemple:
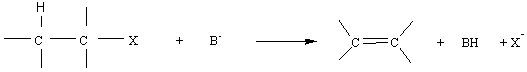
Remarque : réaction
régiosélective : si la double liaison peut se former avec 2 carbones adjacents
qui portent un H, elle se forme de préférence avec celui qui est le plus
substitué (règle empirique de Zaïtsev).
Exemple :
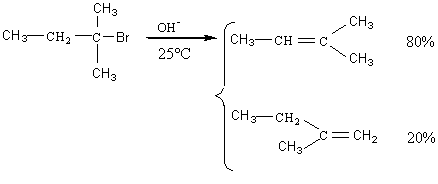
7-2) Elimination
monomoléculaire ou E1 :
1ère étape : la plus lente
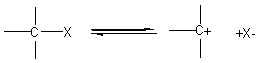
2ème étape : la plus rapide
Vitesse de réaction :
v = k [R-X] ordre 1
Ces réactions ont lieu plus facilement avec des carbones
(III) (carbocations plus stables), avec des bases faibles et des solvants
protiques.
Exemple :
Stéréochimie
:
si la molécule de départ le permet (voir ci-dessous)
le mécanisme E1 conduit à un mélange de stéréoisomères Z et E
- Isomère Z
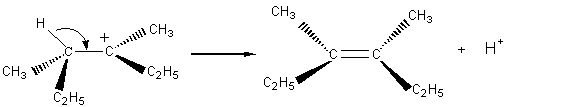
- Isomère E
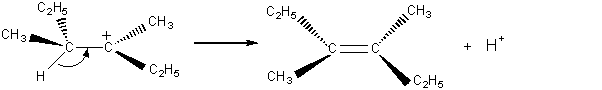
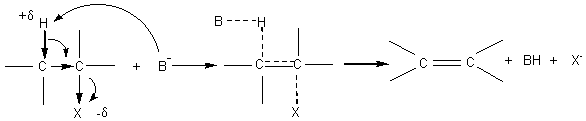
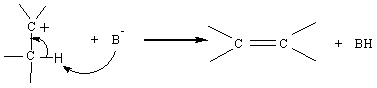
7-3) Elimination
bimoléculaire ou E2 :
Exemple :
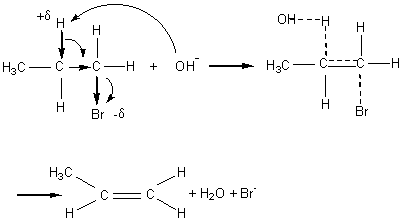
Vitesse :
v = k [RX] [B-]
Ces réactions ont lieu plus facilement avec des carbones
(I), des bases fortes, des solvants aprotiques.
Stéréochimie
:
C' est une anti-élimination (on dit parfois
trans-élimination).
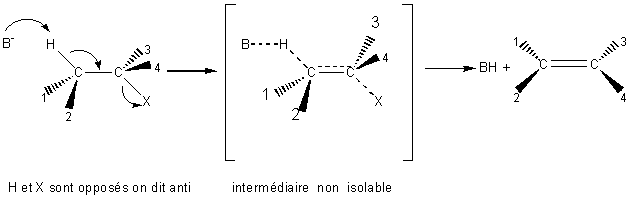
Remarque :Si
les deux carbones sont asymétriques, on obtient un seul stéréoisomère Z ou E et
non un mélange comme dans le mécanisme E1.