LA DISTILLATION (Théorie)
1) Introduction.
- La distillation est la principale méthode de séparation des constituants d'un mélange liquide.
- Cette opération est réalisée en vaporisant le mélange puis en réalisant une succession de condensations et de vaporisations pour terminer par la récupération d'un des constituants.
- L'étude de la distillation nécessite la connaissance des lois des équilibres liquide - vapeur pour les corps purs et pour les mélanges.
2) Equilibre liquide-vapeur d'un corps pur.
2-1) Equilibre liquide vapeur en vase clos - Tension de vapeur.
Un équilibre physique s'établit entre liquide et
vapeur
![]()
La vapeur au-dessus du liquide est dite saturante. Sa
pression p0 est dite :
- pression de vapeur saturante ou
- tension de vapeur.
Cette grandeur p0 est caractérisée
par : p0 = cste à T = cste.
p0 augmente quand T augmente
p0 est indépendante de la quantité de liquide.
Le domaine (Liq + Vap) est représenté par la courbe:
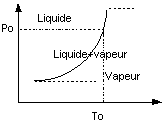
Diagramme pression température ou
courbe de tension de vapeur.
2-2) Equilibre liquide vapeur à l'air libre, à température constante: Evaporation.
- Si le vase est clos: la pression dans le vase p = p0.
- Si on ouvre le récipient : la pression au-dessus du liquide diminue p < p0.
L'équilibre est rompu, pour retrouver p = p0 le liquide émet de la vapeur par sa surface libre c'est le phénomène d'évaporation. Ce mécanisme a lieu tant qu'il reste du liquide. Quand on arrive à l'épuisement du liquide, la vapeur est dite sèche.
- La vitesse d'évaporation augmente : par élévation de la température; par élimination de la vapeur émise.
A température constante
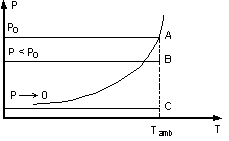
Points représentatifs :
- A récipient fermé pression de la vapeur saturante.
- B ouverture du récipient. La pression de la vapeur
diminue. - C absence de liquide dans le récipient. La pression de vapeur tend
vers 0.
2-3) Vaporisation totale à l'air libre par élévation de température :
Ebullition.
Considérons le liquide dans un récipient ouvert à
l'air libre.
A l'état initial : Température ambiante ; pression de
vapeur p < p0.
- Lorsque l'on chauffe, la température augmente ; p0 augmente ; lorsque p0 = Patm on observe l'ébullition du liquide.
- Ebullition : Vaporisation en surface mais aussi au sein du liquide par émission de bulles de vapeur qui viennent bouillonner à la surface.
La pression de la vapeur émise ne
peut pas augmenter au-delà de la pression imposée Pimp (pression
atmosphérique) car le récipient est ouvert.
Si on chauffe, la quantité de chaleur apportée sert
au changement d'état qui se fait à pression constante (pression atmosphérique)
donc à température constante. Cette température est la température d'ébullition
(Teb) du corps ou point d'ébullition (P.E).
- Remarques :
- Lorsque l'on veut provoquer
l'ébullition d'un corps on peut pour abaisser la température d'ébullition
abaisser la pression (distillation sous pression réduite).
- Une distillation industrielle est souvent pilotée par
une régulation de pression, qui tient compte des variations de la Patm
. Ce système de commande est préféré à la régulation de température.
- Surchauffe :
Phénomène de vaporisation, par évaporation
seulement et absence d'ébullition, s'il n'existe pas un micro volume de gaz au
sein du liquide.
De ce fait la température atteinte T au sein du
liquide est supérieure à la température d'ébullition Teb ;
d'où l'appellation de surchauffe.
En pratique pour éviter cela on place dans le mélange
chauffé un grain de pierre ponce ou quelques billes de verre.
2-4) Condensation à l'air libre par abaissement de température.
Si la
température de la vapeur saturante diminue, cela s'accompagne d'une diminution
de sa pression.
La pression est directement liée au nombre de
molécules de vapeur dans le milieu ; ce nombre doit donc diminuer. Ces
molécules se condensent à l'état liquide.
Dans le cas d'un corps pur la pression de vapeur
saturante p0 = Pimp . La température du début de condensation est appelée
point de rosée à la pression imposée.
3) Equilibre Liquide-Vapeur d'un mélange binaire idéal.
- Mélange idéal : Les espèces qui composent le mélange sont indépendantes les unes des autres.
Nous considérerons un mélange
idéal de (A+B) à l'état liquide en équilibre avec sa vapeur.
Données :
T : Température d'équilibre.
TebAet
TebB :
Températures d'ébullition de A pur et de B pur.
xA et xB : Fractions molaires des composés A et B dans la phase
liquide.
nlAet
nlB :
Nombre de moles de A et de B dans le liquide.
![]()
yA et yB : Fractions molaires des composés A et B dans la phase
vapeur.
nvA et nvB :
Nombre de moles de A et de B dans la phase vapeur.
![]()
Pimp : Pression imposée.
p0A et
p0B : Pressions
de vapeur saturante, à la température T, des composés A et B pris isolément.
pA et pB : Pressions partielles, à la température T, des vapeurs de
A et de B dans la vapeur saturante du mélange (A+B)
p : Pression totale de la vapeur saturante, à la
température T, du mélange (A+B) ; si elle est assimilée à un gaz parfait sa
valeur est p = pA + pB
La loi de Raoult donne la pression partielle de
vapeur, à la température T, de chacun des composés A et B dans le mélange :
pA = p0A .xA et
pB = p0B .xB
La loi des gaz parfaits appliquée à la phase vapeur
permet d'écrire: ![]()
![]()
aA/B : Volatilité relative des deux composés ou facteur
d'enrichissement de la distillation.
Sur un faible intervalle de température aA/B peut être
considéré comme constant.
Si on considère un mélange tel que p0A > p0B ( A est plus
volatil que B) donc aA/B
> 1 ; il vient :
![]() Cette
inégalité montre que la composition de la phase vapeur est plus riche en A
(composé le plus volatil) que la phase liquide.
Cette
inégalité montre que la composition de la phase vapeur est plus riche en A
(composé le plus volatil) que la phase liquide.
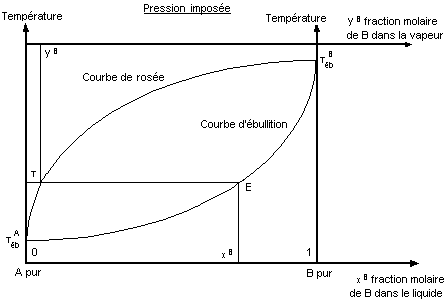
Diagramme
d'équilibre (liq - vap) d'un mélange idéal des composés A et B à la pression Pimp.
- Un mélange liquide (A+B) de composition xB
bout à la température T point (E) de la courbe d'ébullition.
Il émet à cette température, une vapeur de
composition yB , cette vapeur contient moins de B que le liquide qui lui
a donné naissance. La vapeur s'est donc enrichie en composé A (composé le plus
volatil).
La séparation par distillation des constituants d'un
mélange est basée sur ce principe : différence des compositions entre la vapeur
émise que l'on condense (le distillat) et le liquide en ébullition dans le
bouilleur.
4) Principe de la distillation fractionnée d'un mélange binaire idéal.
- Une
distillation est dite fractionnée lorsqu'elle consiste en une suite de
distillations élémentaires réalisées dans un appareil unique. (Cette opération
est souvent insuffisante pour permettre la séparation complète des constituants
du mélange).
Pour obtenir les constituants à l'état pur il faut
procéder à une rectification. (L'appareil présente un plus grand pouvoir
séparateur. L'appareil est parfois surmonté d'un analyseur.)
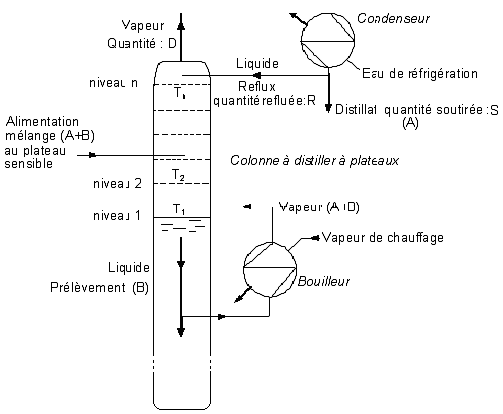
Reflux
ou rétrogradation : C'est le liquide
descendant. Il est toujours provoqué par un système réfrigérant appelé
condenseur ou rétrogradeur ou déflegmateur.
Pour qu'une colonne à distiller puisse jouer son
rôle, il faut un contact entre la phase liquide et la phase vapeur.
Il y a donc nécessité absolue de reflux dans la
colonne.
Taux de reflux
: Deux rapports différents sont acceptés pour définir le taux de reflux.
![]()
avec :
D : débit molaire de la colonne
R : débit molaire reflué
S : débit molaire soutiré
Le bilan matière en tête de colonne donne :
D = R+S. R et S sont facilement mesurables.
Si on ne soutire pas, D = R le reflux est total.
Avec la première définition du taux de reflux t = 1
Avec la seconde t tend vers l'infini .
Dans
l'appareil à distiller :
- Il y a échange de matière et de chaleur entre la phase
vapeur ascendante et la phase liquide descendante. Pour réaliser cet échange,
il faut mettre en contact la vapeur et le liquide.
- Cela se fait par léchage dans le cas des colonnes à garnissage ; l'efficacité est améliorée en augmentant la surface de contact. On introduit en vrac, des petits solides de forme convenable, tels que les vapeurs puissent cheminer dans les interstices au contact du liquide de reflux s'écoulant depuis le haut.
- Cela se fait par barbotage des vapeurs dans le liquide
dans le cas des colonnes à plateaux. Le liquide est retenu sur un plateau
par un système à cloches ou à déversoir.
- La vapeur s'enrichit de plus en plus en constituant le plus volatil, et le liquide en constituant le moins volatil.
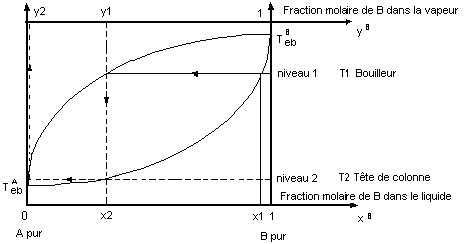
Soit la
distillation d'un mélange idéal A+B de composition x1
placé dans le bouilleur.
Lorsque le mélange est porté à l'ébullition, il émet
une vapeur enrichie en A, qui se condense au niveau 2.
A ce même niveau ce mélange de composition x2
émet une vapeur enrichie en A, qui se condense au niveau 3.
Ce mécanisme se reproduit jusqu'au niveau n du
dernier plateau.
A ce niveau la vapeur émise de composition xn
s'échappe vers le condenseur pour être entièrement condensée.
Une partie de ce condensat est renvoyée vers la
colonne c'est le reflux R (encore appelé rétrogradation); l'autre est prélevée
c'est le distillat S.
Le fonctionnement d'une colonne doit être adiabatique
; ce qui rend nécessaire le calorifugeage de la colonne.
Pour ne pas perturber l'équilibre liquide - vapeur en
tête de colonne et augmenter son efficacité, il faut prévoir un reflux.
5) Caractéristiques générales d'une colonne à distiller.
5-1) Puissance de la colonne.
C'est le nombre de plateaux théoriques de la colonne.
Plateau théorique : c'est, quel que soit le système utilisé, la zone dans
laquelle, liquide à ébullition et vapeur émise sont en équilibre.
Remarques :
Le nombre de plateaux théoriques est repérable par
construction sur le diagramme d'équilibre (liq -vap) du mélange en fonction de
la température.
Sous vide le nombre de plateaux théorique diminue,
donc la colonne est moins puissante.
5-2) Débit.
C'est le volume de vapeur passant dans la colonne
pendant l'unité de temps.
Le débit sera fonction du diamètre de la colonne.
Sous vide on doit diminuer le débit pour conserver le
même nombre de plateaux théoriques.
On ne peut pas augmenter le débit au delà d'une certaine
valeur sous peine de provoquer l'engorgement de la colonne (il se forme
"un bouchon liquide" lorsque les vapeurs empêchent le liquide de
refluer vers le bouilleur).
5-3) H.E.P.T. (hauteur équivalente à un plateau théorique).
On la détermine expérimentalement. Elle varie
largement pour une colonne donnée, en fonction des conditions d'utilisation.
5-4) Retenue de la colonne ou volume de rétention.
- Retenue dynamique : C'est le volume de
rétention en fonctionnement. C'est à dire le volume minimum retenu dans la
colonne et ses parties annexes au cours de la distillation. Il est égal à :
liquide de reflux dans la colonne plus liquide dans le condenseur et le système
de prélèvement, plus liquide correspondant à la vapeur dans la colonne.
- Retenue statique : Liquide retenu dans la
colonne la distillation terminée.
C'est du volume de rétention que dépend la finesse de
la séparation.
5-5) Efficacité.
Rapport du nombre de plateaux théoriques au nombre de
plateaux réels. Pour une colonne donnée elle dépend du taux de reflux et de la
verticalité de la colonne.
5-6) Finesse.
Cette caractéristique permet de comparer des colonnes
ayant même efficacité et même pouvoir séparateur pour un mélange donné. (Elles
permettent d'obtenir le même pourcentage de pureté du produit).
La colonne présentant la plus grande finesse est
celle qui a le plus faible volume de rétention.
5-7) Pertes de charge.
C'est la force que doit vaincre la vapeur pour monter
dans la colonne. Elle a pour origine les obstacles rencontrés par la vapeur
dans son mouvement ascendant ; principalement le liquide de reflux et les
changements de direction liés à la conception de la colonne.
Si cette chute de pression Dp = ppied
de colonne - ptête de colonne est importante elle amènera une
variation de la température d'ébullition dans la colonne.
6) Description d'une colonne à distiller.
- Les
colonnes à distiller ont une forme cylindrique, leur hauteur est toujours très
grande comparée à leur diamètre.
- Les plus utilisées sont équipées de plateaux (ils
peuvent être de plusieurs types); mais il existe aussi des colonnes à
garnissage (là aussi plusieurs types de garnissage sont disponibles sur le
marché).
- Dans le cas des colonnes à plateaux on dispose
régulièrement entre les plateaux des "accès" appelés "trou de
poing" ou "trou d'homme", qui permettent d'entretenir le
matériel sans avoir à démonter l'ensemble de la colonne.
- Les matériaux utilisés peuvent être : l'acier
inoxydable ; le cuivre ; le verre ; ou encore des matières plastiques (pour les
faibles températures).
Remarque :On réalise parfois des
distillations sous vide, le matériel sera le même que pour une distillation
normale mais il faudra : une étanchéité parfaite ; un appareil producteur de
vide ; un appareil régulateur de vide et un autre de mesure du vide. Pour
soutirer le produit une colonne barométrique est obligatoire.
7) Etude des courbes de distillation.
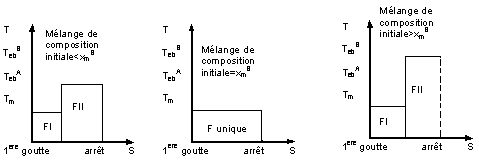
- On regroupe sous cette appellation les courbes représentatives de la température de la vapeur en tête de colonne Tn en fonction du temps ou de la quantité de distillat recueilli, S.
7-1) Solution binaire idéale.
Mélange idéal de deux liquides A et B. A est le plus
volatil.
La distillation du mélange conduit à trois fractions
principales:
- Fraction I : A pur, à la température Tn =
TebA en
tête de colonne.
- Fraction II : Mélange A + B , à la température Tn
telle que TebA
< Tn < TebB .
cette fraction est appelée fraction intermédiaire
entre les composés A et B.
Plus cette fraction (FII) est réduite plus la
séparation est parfaite.
- Fraction III : B pur, à la température Tn =
TebB .
Dans le bouilleur il reste le résidu (B pur) que l'on
ne distille pas pour des raisons de sécurité.
La température T1 dans le bouilleur
est plus élevée que la température Tn en tête de colonne tant
que le composé B ne passe pas seul.
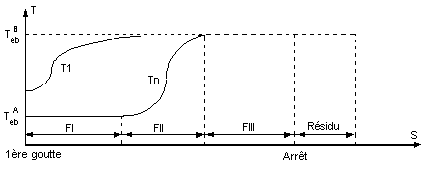
Courbe de distillation d'une solution
idéale température en fonction du nombre de moles soutirées.
7-2)Solutions binaires non idéales - Mélanges azéotropes.
On peut distinguer deux catégories de mélanges non
idéaux :
- Les mélanges proches des solutions idéales ; la
forme du diagramme liquide vapeur est voisin d'un fuseau. Les résultats
précédents sont applicables.
- Les mélanges dits azéotropes. Le nombre et la
nature des fractions obtenues dépendent de la composition initiale du mélange.
Les deux corps purs ne peuvent pas être obtenus par distillation, un seul peut
l’être.
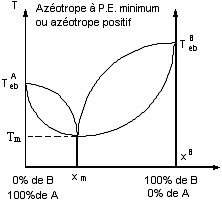
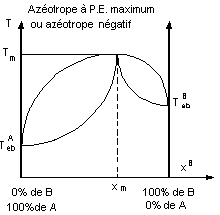
- Les courbes d'ébullition et de rosée passent par un
minimum ou un maximum de température Tm où elles se confondent.
La composition xmB ,
appelée composition azéotropique, a un comportement, en distillation, analogue
à celui d'un corps pur.
- Pour chaque mélange azéotrope, il existe trois
courbes de distillation qui dépendent de la valeur xm de la
composition initiale du mélange.
- Selon la composition initiale du mélange la
distillation conduit à séparer A de l'azéotrope (A+B) ou B de (A+B).
- Pour de tels mélanges la séparation intégrale des
corps A et B est impossible.
- Pour séparer A de B il faudra :
- soit faire une distillation extractive par un tiers
corps ;
- soit distiller sous une pression à laquelle
l'azéotrope n'existe plus ;
- soit fixer chimiquement l'un des constituants.
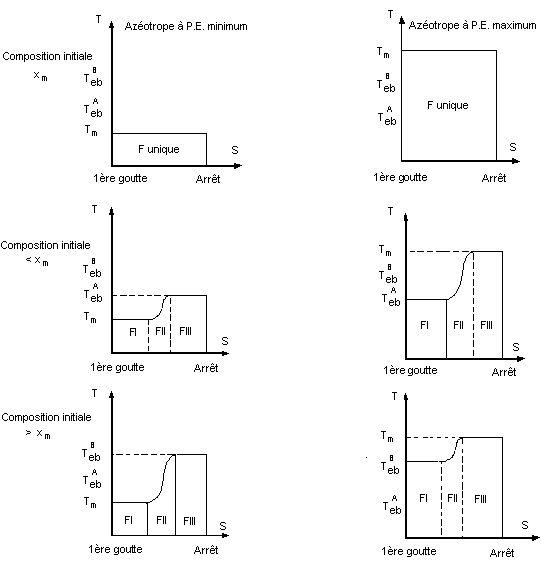
7-3) Mélange de deux liquides partiellement miscibles.
Exemple : eau
aniline. Les courbes de distillation et les conclusions relatives à ce type de
mélange sont analogues à celles obtenues avec un mélange azéotrope à point
d'ébullition minimum.
Ce cas de miscibilité partielle est le plus
fréquemment rencontré ; la composition xmB, appelée hétéroazéotropique a un comportement en
distillation, analogue à celui d'un corps pur.
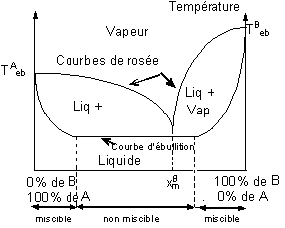
7-4) Mélange de deux liquides non miscibles.
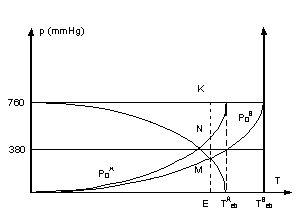
Si on chauffe un mélange de deux liquides A et B non
miscibles, la température d'ébullition du mélange est inférieure à celle du
constituant à point d'ébullition le plus bas. (courbe de gauche ci-dessous).
Quelle que soit la composition du mélange, sa
température d'ébullition est la même et il émet des vapeurs de composition
fixe. (jusqu'à ce que l'un des deux corps soit éliminé).
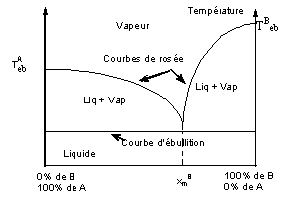
Sur les
courbes de distillation T= f(S), on constate que la nature du distillat dépend
de la composition initiale du mélange distillé.
Interprétation.
- Les deux corps coexistent pratiquement purs l'un en
présence de l'autre. La pression de vapeur de chacun d'eux ne dépend que de la
température. Le mélange bout lorsque sa tension de vapeur p est égale à la
pression supportée. ( Pimp = patm par exemple).
Avec p = p0A + p0B
donc quelle que soit la proportion de A et de B dans le mélange la température
d'ébullition sera inférieure aux points d'ébullition de A et de B.
- La détermination de la température d'ébullition
d'un mélange sou une pression imposée peut se faire à partir des courbes de
tension de vapeur fonction de la température.(courbes de droite ci-dessus).
- Tracer l'horizontale d'ordonnée égale à la demi pression de distillation imposée.
- Tracer le symétrique à l'une des deux courbes de tension de vapeur par rapport à l'horizontale précédente.
- Soit M le point d'intersection des courbes. La verticale passant par M coupe l'axe des abscisses en E. Cette température est la température d'ébullition du mélange. A cette température p0A + p0B = Pimp .
- Composition des vapeurs émises.
La pression partielle de chaque corps dans la vapeur
est proportionnelle au nombre de mole de vapeur de ce corps dans le volume
considéré ; et indépendante de la composition du liquide.
pA = p0A =
k.nvA pB = p0B = k.nvB
La composition
molaire de la vapeur est donnée par : ![]()
La
composition massique de la vapeur :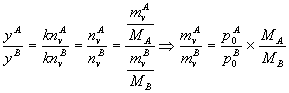
- Le
quotient : ![]() est appelé rapport d'entraînement. Il dépend de la masse
molaire du corps et de sa tension de vapeur.
est appelé rapport d'entraînement. Il dépend de la masse
molaire du corps et de sa tension de vapeur.
- Ce phénomène est mis en oeuvre pour extraire l'un
des constituants, s'il n'est pas miscible à l'eau : c'est l'entraînement à la
vapeur.
- Pour que cette méthode soit justifiable, il faut
que p0A
soit assez importante à la température d'entraînement et que MA
soit aussi de valeur élevée.
- Si le mélange entraîné à la vapeur, contient deux corps
non miscibles à l'eau, ils ne peuvent être entraînés sélectivement que si leur
différence de tension de vapeur est suffisamment importante.
- Les constituants du distillat sont ensuite séparés
par décantation ou par filtration si nécessaire.
8) Applications
de la distillation
Les applications usuelles de la distillation sont les
suivantes :
- élimination d'un produit en cours de réaction
chimique ;
- isolement de plusieurs composés obtenus après
réaction chimique ;
- élimination d'un solvant ;
- isolement d'un composé naturel ;
- purification d'un composé.