LA CHIMIE DURABLE
Synthèse d’une molécule bioactive à
partir de « molécules plateformes » issues de la biomasse
Article bâti autour du sujet de
l'agrégation externe de chimie 2021 dont il contient de larges extraits.
Plan de l'étude
1)
Généralités
2)
Déconnexion rétrosynthétique de l'aspergillide A
3) Obtention des trois
"molécules plateformes"
3-1) Le
bioéthanol
3-2) Obtention
du HMF et de l'acide lévulinique à partir de la biomasse
4) Synthèse de l'aspergillide A
4-1)
Fragment A à partir de l'acide lévulinique
4-2)
Synthèse du fragment B à partir du HMF
4-3)
Assemblage des fragments A et B par couplage de
Negishi en phase micellaire
4-4) Fin
de la synthèse de l'aspergillide A
Annexe 1 Rétrosynthèse
et déconnexion rétrosynthétique
Annexe 2 ADP,ATP, Pi
Annexe 3 Le couplage de Negishi
Annexe 4 Macrolactonisation de Yamaguchi
Annexe 5 transposition d'Achmatowicz
Annexe 6 Iodo-oléfination de Takai
Les énergies fossiles représentent
aujourd’hui plus de 95 % de l’énergie primaire consommée. La combustion des
hydrocarbures fossiles est donnée comme principale source des émissions
anthropiques cumulées de gaz à effet de serre, responsables principaux du
changement climatique déjà amorcé.
Une transition énergétique,
modification structurelle profonde des modes de production et de consommation
d’énergie, est donc nécessaire. Pour ce qui concerne la chimie, une transition
depuis la pétrochimie vers une chimie du carbone renouvelable, c’est-à-dire
issue de la biomasse, majoritairement végétale, est une priorité. Les
recherches scientifiques sur la composition et la transformation chimique de la
biomasse ont permis d’étendre le domaine d’application de cette matière
première initialement tournée vers la valorisation énergétique. En particulier,
la biomasse devient progressivement une source variée de synthons, appelés «
molécules plateformes », utilisables en synthèse totale.
Les « molécules plateformes » sont
des « produits » bruts obtenus à partir des matières premières de la biomasse :
production agricole et forestière (biomasse primaire) ainsi que les déchets et
les produits en fin de vie (biomasse secondaire).
Dans une
bioraffinerie dite "primaire", des étapes de séparation plus ou moins
complexes sont réalisées afin de générer une matière première plus exploitable
pour les procédés chimiques. Ainsi les grands polymères que sont la cellulose,
la lignine et l'hémicellulose sont, par exemple, dissociés par
"fractionnement". Les composés obtenus sont dans la majorité des cas
transformés à l'aide d'étapes biochimiques. On obtient ainsi un ensemble limité
de "molécules plateformes", correspondant aux "molécules de
commodités" d'une pétroraffinerie. Ces molécules sont ensuite converties,
raffinées, formulées dans la bioraffinerie secondaire, dans laquelle la chimie
au sens large (catalyse, biocatalyse, thermochimie, etc.) possède un rôle
majeur. Des arbres de produits sont ainsi développés pour aboutir in fine à
des produits finis aux propriétés d'usage ciblées, répondant à des objectifs
commerciaux et de rentabilité de la filière.
Le
tableau ci-dessous recense ainsi un ensemble de "molécules
plateformes" de C1 à C6. Elles sont pour la plupart dérivées du glucose,
lui-même pouvant être issu de la cellulose.
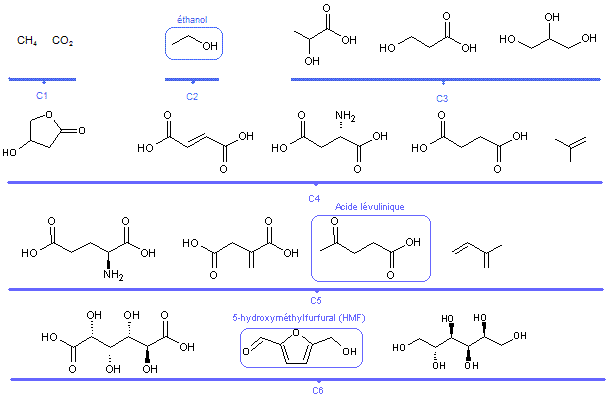
Les
molécules "plateformes" constituent des objets d'études extrêmement
intéressants dans le cadre de développements méthodologiques en synthèse organique,
notamment lorsqu'il s'agit de développer des molécules aux fonctionnalités
nouvelles destinées à des secteurs à moyenne ou haute valeur ajoutée tels que
l'industrie pharmaceutique, cosmétique ou encore agrochimique.
On va
étudier une synthèse organique totale d'une molécule complexe, l'aspergillide A
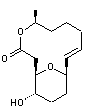
Cette
synthèse d'un produit naturel possédant une activité biologique très
prometteuse, a été réalisée de sorte que tous les atomes de carbone présents
proviennent de "molécules plateformes" issues de la biomasse.
Ce type
de synthèse utilisant l'ensemble des "molécules plateformes" issues
de la biomasse à la place des réactifs chimiques provenant de ressources
fossiles connaît un développement important. Tout cela dans un esprit de
durabilité mais aussi d'économie potentielle.
2) Déconnexion rétrosynthétique de l'aspergillide A (voir annexe 1)
La
déconnexion rétrosynthétique des l'aspergillide A est représentée ci-dessous :
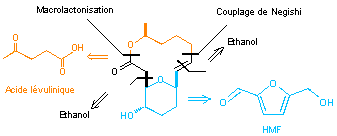
Les
"molécules plateformes" utilisées pour sa synthèse sont l'acide
lévulinique, le 5-hydroxyméthylfurfural (HMF) et l'éthanol.
3) Obtention des trois "molécules plateformes"
L'éthanol
obtenu à partir de la biomasse est généralement appelé "bioéthanol".
Tous les sucres fermentescibles possédant 6 carbones, principalement le glucose,
peuvent être convertis en éthanol et en dioxyde de carbone après fermentation.
Celle-ci s'effectue de manière anaérobie et est catalysée par une enzyme
produite par une levure "Saccharomyces cerevisiae".
L'équation
chimique simplifiée de la réaction modélisant la fermentation anaérobie du
glucose solide en éthanol liquide catalysée par Saccharomyces cerevisiae s'écrit
(voir annexe 2)
![]()
Le
bioéthanol sert principalement d'additif aux carburants traditionnels (essence et gazole), mais il devient
également une "molécule plateforme" essentielle pour la chimie des
grands intermédiaires biosourcés comme le "bioéthylène" ou le
"biobutadiène" permettant de de fabriquer des
"biopolymères".
On
parle couramment de "bioéthanol" de première et de deuxième
génération et même de troisième génération.
La
différence entre ces trois générations réside dans la matière première utilisée
dont dépendent les techniques mises en oeuvre pour obtenir l'éthanol.
- "Bioéthanol" de
première génération
On
soumet les jus sucrés provenant des cannes à sucre, des betteraves sucrières,
des céréales … à une fermentation. Pour l'heure c'est de loin la technique la
plus exploitée industriellement. Cette production entre en concurrence avec la
chaîne alimentaire.
- "Bioéthanol" de
deuxième génération
Exploitation des matières cellulosiques ou
ligno-cellulosiques non utilisées dans la production alimentaire. Les
développements industriels sont pour l'instant limités.
- "Bioéthanol" de
troisième génération
Certaines
microalgues contiennent des sucres qui peuvent être fermentés et qui peuvent
conduire à du "bioéthanol". Cette voie est au stade de la recherche
actuellement.
3-2) Obtention
du HMF et de l'acide lévulinique à partir de la biomasse
Parmi l'ensemble des "molécules
plateformes", le 5-hydroxyméthylfurfural (HMF)
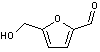
et l'acide lévulinique
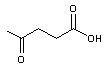
tiennent une place majeure
puisqu'elles ont été répertoriées par le département de l'Energie des
Etats-Unis (DoE) comme deux des douze molécules à produire prioritairement par
les bioraffineries.
En effet, le HMF est un
intermédiaire à forte valeur ajoutée qui peut, par exemple, être oxydé en acide
2,5-furanedicarboxylique (FDCA),
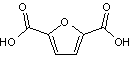
monomère de choix dans la synthèse
d'un polyester 100% biosourcé, le poly(furanoate d'éthylène), nommé PEF,
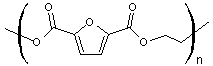
qui peut être utilisé pour la
fabrication de bouteilles (à la place du polytéréphtalate d'éthylène, PET)
Le HMF est également le précurseur
de l'acide lévulinique, qui, par estérification avec les alcools légers,
fournit des solvants. Cet acide a aussi été utilisé comme intermédiaire dans la
synthèse de résines thermodurcissables et comme synthon pour la préparation de
principes actifs.
Le glucose et le fructose sont les
deux précurseurs classiquement utilisés pour obtenir du HMF. Par triple
déshydratation à hautes températures et en présence d'un catalyseur acide, ces
deux monosaccharides sont transformés en HMF. Les rendements en HMF sont
meilleurs lorsque le substrat est le fructose car il possède déjà une structure
cyclique à cinq chaînons (appelée furanose) qui est la même que celle du HMF.
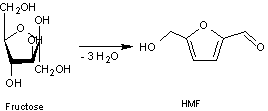
Remarque : Le HMF peut ensuite réagir sur l'eau et se décomposer en
acide lévulinique et en acide formique
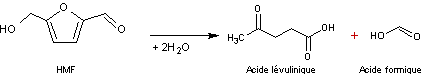
Le rendement de la conversion du
fructose en HMF passe par un maximum avant de diminuer du fait de la
dégradation du HMF en acides lévulinique et formique ; il a donc été envisagé
d'extraire en continu le HMF formé par du dioxyde de carbone supercritique. En
plus de son pouvoir extractant, le dioxyde de carbone sert à acidifier la phase
aqueuse ce qui catalyse la synthèse du HMF.
4) Synthèse de l'aspergillide A
L'aspergillide A est un macrolide cytotoxique produit par le champignon
marin Aspergillus ostianus. Cette molécule a été isolée en 2008 par le
groupe de Kusumi. Ce composé présente une activité cytotoxique puissante contre
les cellules de leucémie lymphoïdes de souris (L1210) avec une valeur de LD50
de seulement 2,1 µg.mL-1.
De plus, les architectures moléculaires
uniques des aspergillides ont attiré une attention particulière de la part de
la communauté de la chimie de synthèse car ce sont les premiers exemples de
structure macrocyclique à 14 atomes incorporant une unité tétrahydro ou
dihydropyrane sous une forme qui n'est pas un hémiacétal.
On commence par faire la synthèse de
deux fragments principaux A et B, deux synthons qui sont ensuite assemblés pour
donner l'aspergillide A.
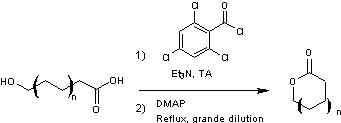
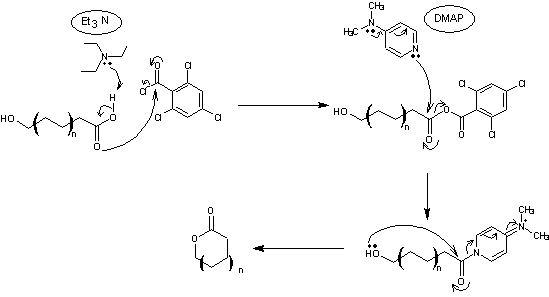
4-1) Fragment A à partir de l'acide lévulinique
L'acide lévulinique est d'abord
réduit en un diol, le pentan-1,4-diol (mélange racémique)
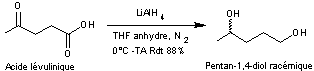
Le mélange obtenu est ensuite soumis
à un dédoublement cinétique enzymatique avec une lipase (Novozyme® 435) en présence
d'acétate de vinyle, à température ambiante (TA) pendant 4 heures.
L'alcool primaire est acétylé le
premier ; il s'ensuit une acétylation énantiosélective de la fonction alcool
secondaire de l'un des énantiomères (le (R)).
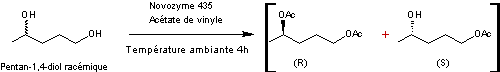
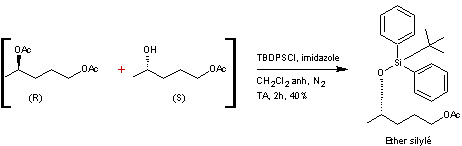
Le mélange brut est directement
repris dans le dichlorométhane puis de l'imidazole et du chlorure de
ter-butyldiphénylsilane (TBDPSCI) sont ajoutés. Le mélange obtenu est agité
pendant deux heures avant traitement et purification pour donner un éther
silylé.
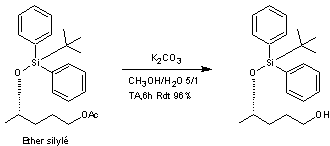
L'éther silylé est traité par du
carbonate de potassium dans un mélange hydroalcoolique (eau-méthanol).
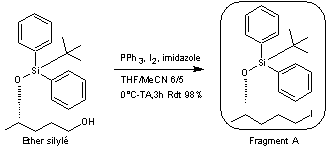
Le composé obtenu est ensuite traité
par 2,0 équivalents de triphénylphosphine, 4,0 équivalents d'imidazole et 2,2
équivalents de diiode dans un mélange THF/acétonitrile (6/5) à température
ambiante pendant 3 heures pour conduire, avec un rendement de 98% au fragment A
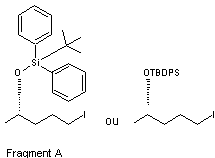
le fragment A peut se représenter de
deux façons :
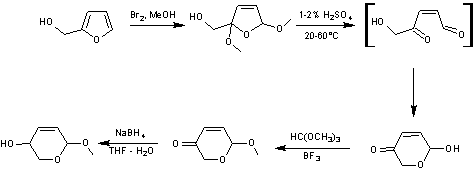
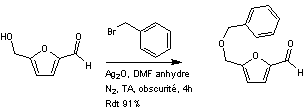
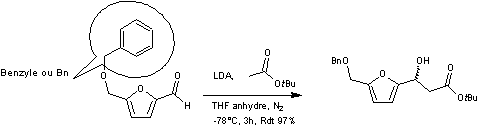
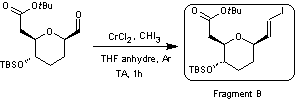
4-2) Synthèse du fragment B à partir du HMF
La première étape de la synthèse de
ce fragment consiste à protéger l'alcool primaire du HMF sous forme d'éther
oxyde benzylique
De l'acétate de ter-butyle (1,5 éq) dans
le THF anhydre (20mL) est ajouté lentement à une solution de diisopropylamidure
de lithium (LDA) (1,5 éq) dans le THF anhydre à -78°C, puis le mélange est
agité pendant 30 minutes. Le HMF dont la fonction alcool a été protégée et qui
a été préalablement dissous dans le THF anhydre (45 mL) est alors ajouté
goutte-à-goutte toujours à – 78°C. Le mélange est agité pendant encore 3 heures
à cette même température. On obtient un β-hydroxyester racémique
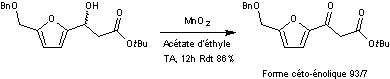
Le mélange racémique est mis à
réagir avec le dioxyde de manganèse pour obtenir par oxydation de la fonction
alcool, un mélange céto-énolique dans les proportions 93/7.
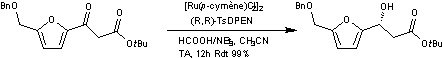
Par un transfert d'hydrogène
asymétrique de Noyori on obtient un mélange non racémique.
Les transferts d'hydrogène sont des méthodes
douces de réduction des cétones (ou d'imines) dans lesquels le catalyseur
sélectif du substrat transfère de l'hydrogène entre le substrat et un donneur.
Noyori a développé des complexes
chiraux énantiopurs qui se révélèrent très efficaces pour le transfert
d'hydrogène sur des cétones conjuguées avec des systèmes aromatiques pour
donner l'alcool correspondant avec une grande pureté énantiomérique. Ces
travaux lui ont valu l'attribution du prix Nobel de chimie en 2001.
Cette synthèse débute par la préparation
du catalyseur C :
On mélange le complexe dimère
dichloro(p-cymène)ruthénium(II) commercial (263 mg, 0,43mmol, 2,5 mol%),
la (1R,2R)-N-p-tosyl-1,2-diphényléthylènediamine (R,R)-TsDPEN (376 mg, 1,03 mmol, 6mol%) et
MeCN (34 mL) avant d'ajouter de la triéthylamine (356µL, 260mg, 2,57mmol,
15mol%). Le mélange est laissé sous agitation pendant 1 heure à température
ambiante. Le complexe C de couleur orange foncé est obtenu.
Un mélange d'acide méthanoïque et de
triéthylamine en proportions 5/2 v/v (8,6 mL) est ajouté au complexe C puis le
mélange obtenu est ajouté au composé à oxyder (5,66g, 17,2 mmol, 1,0 éq) avant
d'être laissé sous agitation pendant 1 heure à température ambiante pendant 12
heures. Le mélange est alors concentré sous pression réduite et purifié par
chromatographie sur colonne de silice (éluant : hexane/acétate d'éthyle 4/1)
pour conduire à une huile jaune pâle (5,66g, 17,0 mmol, Rdt 99%, ee = 98%). Le
composé avec le centre stéréogène de configuration (R) est majoritaire.
En faisant subir au composé
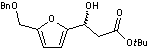
une transposition d'Achmatowicz
(voir annexe 5) suivie d'une triple réduction on obtient
deux pyranes
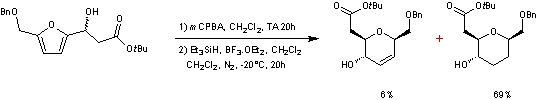
Les deux composés du mélange
réactionnel sont protégés sous forme d'éthers de ter-butyldiméthylsilyle
pour donner un mélange inséparable de deux composés nouveaux
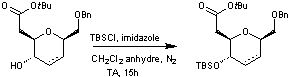
On enlève ensuite le groupe benzyle
protecteur de la fonction alcool primaire en même temps qu'on hydrogène la double
liaison d'un des composés pour n'obtenir qu'un seul produit avec un rendement
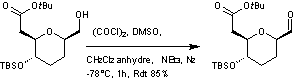
de 96%.
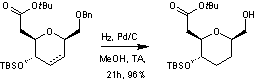
L'alcool obtenu est traité par un mélange
de chlorure d'oxalyle (1,5 éq) et de diméthylsulfoxyde (3,0 éq) dans le
dichlorométhane à – 78°C ; on additionne ensuite de la triéthylamine (6,0 éq).
On extrait et on purifie le produit obtenu.
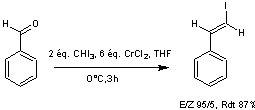
L'iodo-oléfination de Takai (voir annexe 6) appliquée au composé obtenu permet d'obtenir le
fragment B sous forme d'un mélange de deux diastéréoisomères E/Z (81%/19%)
séparables.
Après séparation par chromatographie
sur colonne de silice, le produit majoritaire est isolé et sa configuration
confirmée par analyse cristallographique aux rayons X sur un monocristal du
produit.
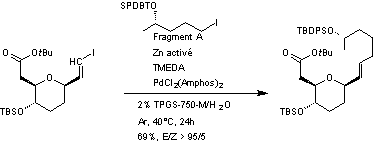
4-3) Assemblage des fragments A et B par couplage de
Negishi (voir annexe
3) en phase micellaire
Les deux fragments A et B formés sont associés
par couplage de Negishi en phase micellaire en utilisant un surfactant approprié
(TPGS-750-M)
Ces conditions opératoires pour
réaliser la réaction de Negishi offre plusieurs avantages comme, par exemple
l'utilisation de l'eau comme solvant de réaction. Par ailleurs, elle ne
nécessite pas de préparer l'organozincique en amont. Après optimisation, les
deux fragments A et B sont couplés pour obtenir un composé comme indiqué
ci-dessous avec un rendement de 69% et une excellente sélectivité E/Z de plus
de 95/5.
Les micelles se forment à partir
d'une certaine concentration en tensioactif appelée concentration micellaire
critique (CMC), généralement comprise entre 10-5 et 10-2
mol.L-1. Cette valeur de la CMC dépend essentiellement de la
longueur de la chaîne hydrocarbonée, du degré d'insaturation et de ramification
du tensio-actif.
4-4) Fin de la synthèse de l'aspergillide A
-
Une stratégie de protection/déprotection
Le groupe éther de triméthylsilyle
du composé obtenu est ensuite clivé par
utilisation du fluorure de tétra-n-butylammonium (TBAF) avant de reprotéger
l'alcool obtenu en éther de méthoxyméthyle (MOM).
On procède ensuite à une hydrolyse
dans des conditions basiques pour conduire à un hydroxyacide.
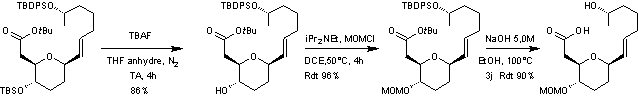
-
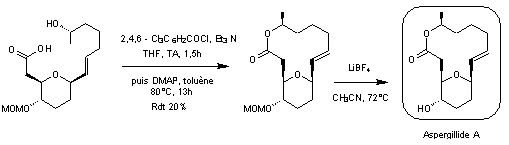
Macrolactonisation de Yamaguchi (voir annexe 4)
L'hydroxyacide obtenu subit une
macrolactonisation selon le protocole suivant :
À une solution de l’acide
carboxylique (13,3 mg ; 0,0420 mmol) dans le THF (5,0 mL) refroidie à 0°C sont
ajoutés de la triéthylamine Et3N (0,045mL ; 0,32 mmol) et du
chlorure de 2,4,6-trichlorobenzoyle (0,035 mL ; 0,22 mmol). Un premier
intermédiaire est formé. Le mélange obtenu est alors agité à température
ambiante pendant 1,5 heure. Le mélange est dilué avec du toluène (70 mL) puis
ajouté à une solution de 4-diméthylaminopyridine (DMAP) (770 mg; 6,30 mmol)
dans le toluène (140 mL) sur une période de 13 heures. Un deuxième
intermédiaire est formé avant de conduire au produit final. Le mélange est
alors refroidi à température ambiante et lavé avec successivement de l’acide
chlorhydrique à 0,5 mol.L–1 , puis avec une solution aqueuse saturée en
hydrogénocarbonate de sodium et enfin avec une solution de saumure. La phase
organique est séchée sur du sulfate de sodium anhydre, filtrée puis concentrée
sous pression réduite. Une purification par chromatographie flash sur gel de
silice (10 à 20% éther diéthylique/benzène) donne la macrolactone (2,5 mg, 20%) sous forme d’une huile
incolore.
Rétrosynthèse
et déconnexion rétrosynthétique
A partir
de la molécule à synthétiser on envisage des entités simples (appelées synthons)
qui en réagissant entre elles permettront d'arriver au résultat. Cette démarche
constitue une "analyse rétrosynthétique".
Pour
identifier ces molécules simples il faut envisager de déconnecter la molécule
d'arrivée en certains points chimiquement stratégiques. Ces déconnexions
constituent la "déconnexion rétrosynthétique".
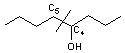
Exemple :
On veut
synthétiser l'octan-4-ol à partir de deux fragments plus petits.
![]()
La position la plus favorable pour créer une liaison C-C (position stratégique)
semble être C4-C5
La
déconnection rétrosynthétique s'écrit :
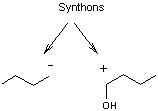
On peut,
pour fabriquer cette molécule penser à deux synthons :
L'analyse
rétrosynthétique s'écrit
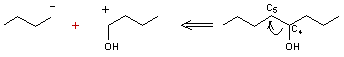
Il faut ensuite
trouver des équivalents réels aux deux synthons, qui en réagissant l'un sur
l'autre donneront la molécule ; en résumé :

ADP,ATP, Pi

Le
phosphate inorganique (Pi) correspond à l'ion phosphate PO43-
Le couplage de Negishi
Le
couplage de Negishi peut se résumer ainsi :
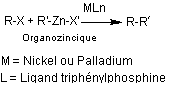
Ces
réactions ont permis la synthèse de composés importants ; elles ont permis par
exemple la synthèse totale de la diazonamide A substance permettant
l'inhibition de l'assemblage de la tubuline et utilisée notamment contre les
tumeurs du côlon.
Negishi
obtient le Prix Nobel de chimie conjointement avec Heck et Suzuki en 2010 pour la mise au
point de synthèses par couplage croisé catalysées par le Palladium.