REACTIVITE DE LA DOUBLE LIAISON CARBONE-CARBONE
Plan de la leçon:
1) Réactivité des doubles liaisons Carbone-carbone
2) Addition
électrophile
3) Addition radicalaire
1) Réactivité des doubles liaisons carbone-carbone :
Une double
liaison carbone-carbone dans une chaîne carbonée, par la densité d'électrons
qu'elle entraîne, est un site, souvent "attaqué" par des réactifs
électrophiles.
Elle est constituée d'une liaison s
forte, et d'une liaison p plus faible;
les électrons de cette liaison p sont
plus mobiles, elle est donc plus polarisable; elle s'ouvre assez facilement et
donne lieu à des réactions d'addition électrophiles.
2-1) réactif dissymétrique A-B :
- Bilan :
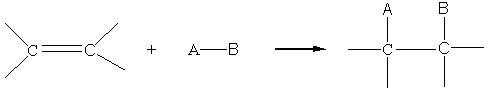
- Mécanisme :
o 1ère étape : il y a attaque de la double liaison par le côté électrophile de A-B. Cette étape sera cinétiquement limitante ...c'est pourquoi on donnera à ces réactions le nom d'additions électrophiles.
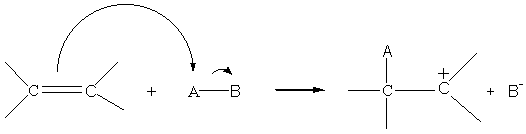
o 2ème étape : il y a addition nucléophile de B-
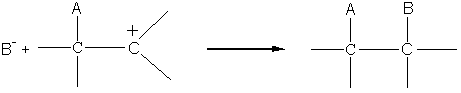
- Bilan:
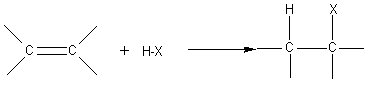
A-B est H-X (halogénure
d'hydrogène) ;
- Mécanisme:
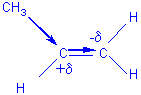
La polarisation de la molécule![]() du fait de l'électronégativité de X, nous montre quelle est la partie de la
molécule qui se montre électrophile (H).Si l'environnement de la double liaison
n'est pas symétrique, il y a deux possibilités d'addition:
du fait de l'électronégativité de X, nous montre quelle est la partie de la
molécule qui se montre électrophile (H).Si l'environnement de la double liaison
n'est pas symétrique, il y a deux possibilités d'addition:
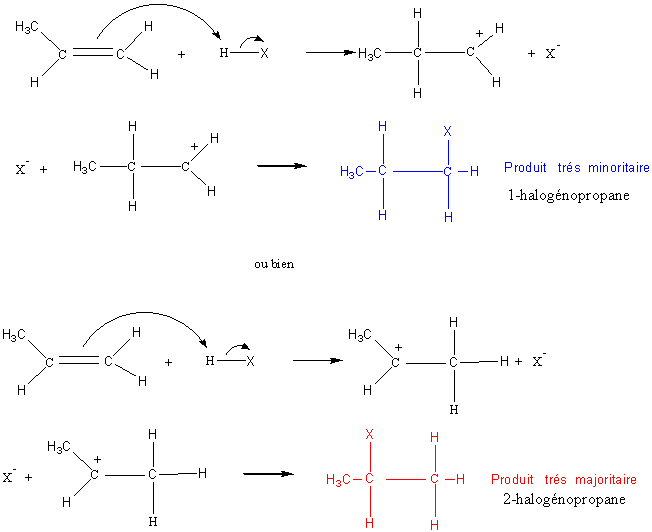
Cette addition est régiosélective ;
elle se fait préférentiellement avec fixation
de H sur le carbone le moins substitué (règle empirique de MARKOVNIKOV).
Cela s'explique par l'effet inducteur répulsif
du groupement CH3- qui
"déplace" le doublet de la liaison p
vers le carbone le moins substitué, le rendant plutôt négatif et facilitant de
ce fait l'attaque de l'extrémité électrophile de la molécule de HX.
Remarque1 :Envisageons le diagramme
énergétique de ces réactions:
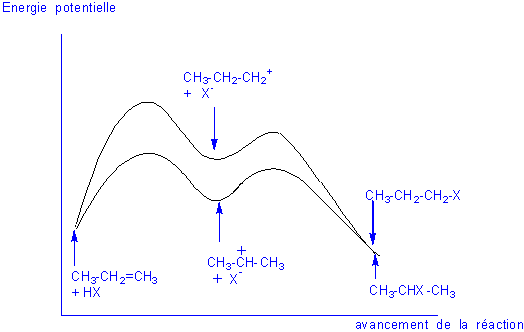
On voit que non seulement le carbocation
secondaire est plus stable ("creux d'énergie" plus important),
mais que l'énergie d'activation (énergie nécessaire pour
"franchir le col" qui conduit au carbocation, en partant des
réactifs) est plus faible, pour le carbocation le plus substitué.
Les 2 considérations, thermodynamique (stabilité) d'une part, et cinétique
(vitesse plus grande car énergie d'activation plus faible) d'autre part,
expliquent la règle empirique de Markovnikov.
Remarque2 : différence
entre régiosélectivité et régiospécificité:
Une réaction régiospécifique, ne conduit qu' à une molécule et une seule ; Une
réaction régiosélective conduit surtout à une molécule, mais sans exclure
totalement la formation d'une petite quantité de l'autre.
Exemple 2 : Hydratation
- Bilan:
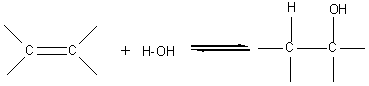
A-B est H-OH (H2O, l'eau).
- Mécanisme:
La différence avec H-X, est que H-OH ne peut protoner la double liaison, pour donner le carbocation ; c'est un acide trop faible. Il faut donc faire intervenir un acide protonique, en général H2SO4( qui a l'avantage sur les hydracides, d'avoir un anion associé, HSO4- moins nucléophile que X- et qui réagira certes sur le carbocation formé, mais qui s'effacera facilement devant un nucléophile plus puissant comme H2O par exemple).
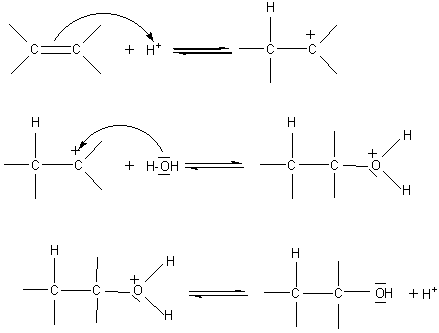
Remarque 1:Cette addition est conforme à la règle de Markovnikov, dans le cas d'un alcène dissymétrique.
Remarque 2:Si le produit de départ est un alcène, on aboutit à un alcool saturé.
Remarque 3:Le H+
qui avait été fourni par l'acide sulfurique à la première étape, est régénéré à
la dernière étape ; H2SO4joue donc le rôle d'un catalyseur.
Toutes les étapes du mécanisme sont des réactions réversibles; H2SO4
devra être utilisé dilué et à basse température (rappelons que concentré et à
haute température, il favorisera au contraire, l'élimination d'une molécule
d'eau d'un alcool).
2-2) réactif symétrique A-A :Halogénation
A-A est X-X (un halogène) : La polarisabilité de la molécule X2 à l'approche d'un site riche en électrons (double liaison) explique l'apparition d'une extrémité électrophile de la molécule d'halogène.
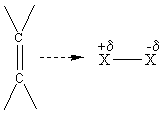
puis formation d'un ion halogénium (X+ se met en pont entre les 2
carbones).

Il s'agit d'une réaction stéréospécifique
Une réaction stéréospécifique appliquée à deux
diastéréo-isomères aboutit à des produits différents.
Prenons l'exemple de l'addition électrophile de
Br2 sur un alcène. C'est une addition anti. On donne ci-dessous le
mécanisme de la réaction :
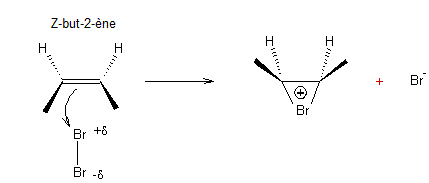
puis

Les deux attaques par l'ion Br- ayant
la même probabilité de se produire, on obtient les deux énantiomères en
quantité égale c'est-à-dire le mélange racémique.
En partant de l'isomère (E) on aboutit aux
stéréo-isomères (2S,3R) et (2R,3S) c'est-à-dire, ici, à la même molécule, le
composé méso.
Remarque : Cette addition ne fonctionne bien qu'avec Cl2 et Br2 (explosive avec le fluor, elle n'a pas lieu avec le diiode).
Hydrohalogénation ( en présence de peroxydes ou de lumière, par exemple) :
c'est une réaction en chaîne, avec 3 phases classiques :
- initiation
:
- propagation :
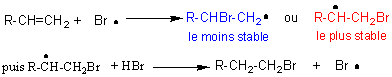
la réaction peut ainsi se poursuivre avec le radical ainsi régénéré.
- fin : elle peut avoir lieu de plusieurs façons:
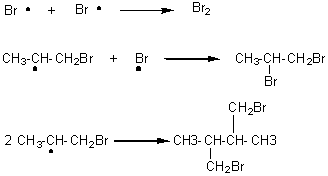
Remarque : contrairement à l'addition électrophile de H-X sur la double liaison, qui conduisait au composé majoritaire CH3-CHX-CH3, le composé majoritaire par cette addition radicalaire est CH3-CH2-CH2Br.
Cette addition est
anti-Markovnikov (c'est l'effet Kharasch).
Remarque: Cette addition radicalaire n'a lieu qu'avec HBr (la dissociation homolytique de HCl demande trop d'énergie; avec HI, la dissociation a lieu, mais la fixation de I- se fait mal).