|
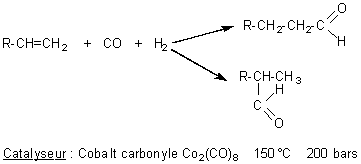
ADKINS (oxosynthèse)
Passage d'un alcène à un aldéhyde avec allongement de la chaîne.
L'addition de monoxyde de carbone en présence de dihydrogène
conduit à un aldéhyde avec allongement de la chaîne.
Cette réaction comparable à celle de Reppe utilise le cobalt carbonyle comme catalyseur.
Cette réaction porte le nom d'oxosynthèse
ou d'hydroformylation
(Tout se passe comme s'il y avait addition de méthanal aussi appelé
formaldéhyde).
En 1975 un procédé de catalyse homogène a été mis au point ; la
préparation du catalyseur est la suivante : du rhodium sous forme d'hydrure
carbonylé, RhH(CO)4 est mis en présence d'un ligand, la triphénylphosphine (TPP), pour former le complexe
rhodium-TPP ; ce dernier se présente sous la forme RhHCO(TPP)3 et
se trouve être très soluble dans le milieu réactionnel organique, dans les
conditions de la réaction qui a lieu dès 20°C et sous faible pression.
|
|
ADKINS
Réduction d'un acide carboxylique en alcool primaire.
Un acide carboxylique peut être réduit en alcool primaire par le
dihydrogène sous une pression de 200 bars à 300°C en présence de chromite de
cuivre comme catalyseur.
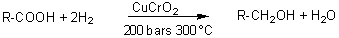
La réduction peut aussi se faire en présencede
LiAlH4 ; la méthode ne s'appelle plus alors méthode d'Adkins.
|
|
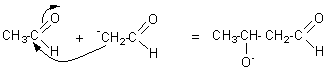
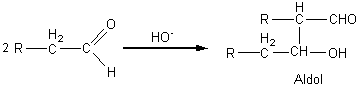
ALDOLISATION
En milieu basique (OH-)
2 molécules d'aldéhydes peuvent s'additionner pour
donner un aldol ( Cette réaction
n'évoluera pas après formation de l'aldol si elle est menée à basse
température (5°C); A température plus élevée, il se
produira une crotonisation
de l'aldol).
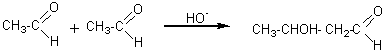 (3-hydroxybutanal) (3-hydroxybutanal)
Mécanisme:
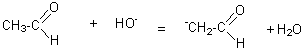
Remarque: cette étape est l'étape lente de la réaction.
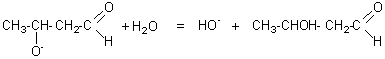
Le Français Wurtz et
le Russe Borodine sont à l'origine de cette réaction.
|
|
ANALYSE
RETROSYNTHETIQUE (principe)
L'idée de base est de
partir de la molécule M0 qu'on souhaite préparer
pour élaborer une voie de synthèse.
On cherche dans les
bases de données la (ou les) molécule(s) M1 permettant en une étape d'obtenir M0 puis la (ou les)
molécule(s) permettant d'obtenir M1et ainsi de suite. Parmi les
voies trouvées le chimiste doit ensuite choisir, en fonction du prix de
revient, donc du rendement global de la chaîne de réactions....
Exemple :On veut synthétiser l'octan-4-ol à partir de
deux fragments plus petits par exemple à 4 carbones.

La position la plus
favorable pour créer une liaison C-C (position stratégique) semble être
C4-C5, car on sait qu'un organomagnésien peut agir sur un aldéhyde pour
donner un alcool.
L'analyse rétrosynthétique de cette synthèse va s'écrire:
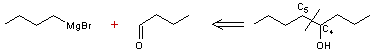
Cet exemple est une
synthèse à une étape, la plupart du temps, plusieurs étapes sont nécessaires.
C'est Corey Professeur à Harvard et
Prix Nobel 1990 qui a conçu cette technique.
Notion de synthons
(building blocks):
Corey
a introduit la notion de synthon qui correspond à une partie fictive de la
molécule à synthétiser qui a des équivalents réels ; l'un de ces équivalents
réels peut réagir avec un des équivalents réels d'un autre synthon pour
donner la molécule recherchée.
Dans
l'exemple de l'analyse rétrosynthétique de
l'octan-4-ol ci-dessus on peut, pour fabriquer cette molécule penser à deux
synthons :

|
|
ARBUZOV
La réaction d'Arbuzov, parfois appelée
réaction de Michaelis-Arbuzov permet d'obtenir un alkylphosphonate à partir d'un trialkylphosphite
:
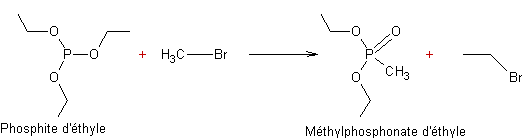
Mécanisme :

|
|
ARNDT-EISTERT
La synthèse de Arndt-Eistert permet de passer
d’un acide à son terme supérieur : -CO2H → -CH2CO2H
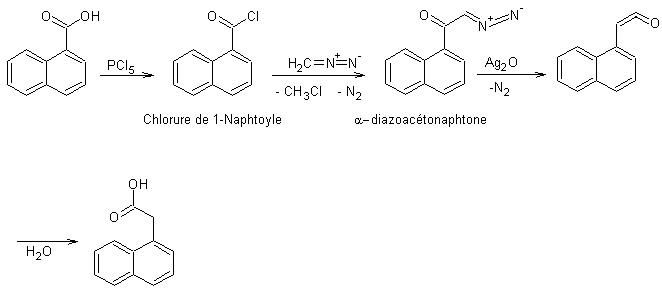
Cette réaction d’un chlorure d’acyle sur le diazométhane
conduit à une diazocétone, qui engendre un cétène
en présence de Ag2O. L’action de l’eau sur ce cétène produit un
acide.
Remarques :
1) L’action du cétène
formé (R-CH=C=O) sur un alcool produit un ester ; par action sur une
amine, il produit un amide.
2) Le diazométhane se prépare à partir de nitroso-amides.
Ceux-ci en milieu alcalin s’isomérisent en diazo-esters
puis se scindent :
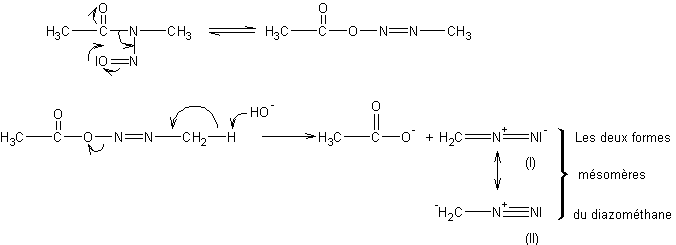
|
|
BAEYER-VILLIGER
Il s'agit de
l'oxydation d'une cétone par un acide peroxycarboxylique
; après addition de l'acide qui aboutit à l'analogue peroxydé d'un
hémiacétal, il y a transposition cyclique qui conduit à un ester.
1èreétape: addition
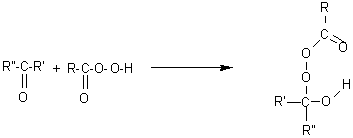
2èmeétape : Transposition
cyclique
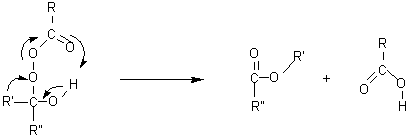
Ainsi la butan-2-one(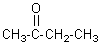 ) devient l'éthanoate d'éthyle( ) devient l'éthanoate d'éthyle(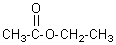 ) )
Lequel des deux
groupes R' ou R" migre ? C'est le plus substitué (C tertiaire migre plus
facilement que C secondaire, lui même plus
facilement que C primaire.)
Les agents oxydants
les plus communément utilisés sont : l'acide perbenzoïque
C6H5CO3H, l'acide peracétique
CH3CO3H, l'acide trifluoroperacétique
CF3CO3H.
|
|
TRANSPOSITION DE BECKMANN
Il s'agit du réarrangement
de l'oxime des cétones qui permet souvent une synthèse des amines:

Les oximes possèdent
deux isomères du type Z/E. C'est le groupe R- antiparallèle avec OH qui migre
du carbone sur l'azote.
L'oxime de la cyclohexanone conduit au caprolactame qui sert à
préparer des fibres textiles artificielles (perlon):
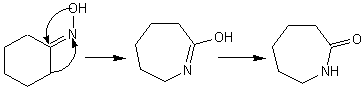
A partir d'un aldoxime (R remplacé par H) on obtient un nitrile
(R'-CN).
Rappel:Les oximes sont des composés cristallisés obtenus
par action de l'hydroxylamine NH2OH sur les composés carbonylés:

|
|
REACTION DE BOUVEAULT ET BLANC
Il s'agit d'une
réaction de réduction des esters en alcool primaire par le sodium en milieu
alcoolique (éthanol).
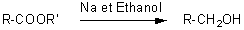
|
|
REACTION DE CANNIZZARO
Dismutation d'un aldéhyde aromatique placé en milieu
basique.
Les aldéhydes aromatiques, le méthanal et tous les
aldéhydes sans H en a ne peuvent donner des réactions d'aldolisation
En milieu basique deux
molécules donnent lieu à une réaction de dismutation
(l'une est oxydée, l'autre est réduite) appelée réaction de Cannizzaro:
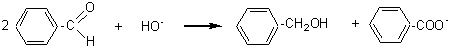
Le mécanisme est
le suivant :

|
|
REARRANGEMENT DE CARROLL
En milieu basique, un ester β-cétoallylique
se transforme en un acide carboxylique, allylique en α et cétonique en
β. Une décarboxylation s’ensuit et l’on aboutit finalement à une γ,d-allylcétone.
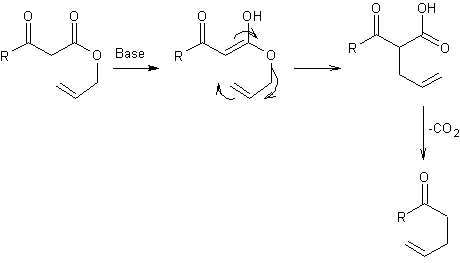
|
|
CETOLISATION
Condensation d'une
molécule d'aldéhyde et d'une molécule de cétone.
En milieu basique une
molécule de cétone et une molécule d'aldéhyde ou deux molécules de cétone (le rendement
dans ce cas est très faible) donnent une réaction comparable à l'aldolisation. C'est l' H en a du groupement
carbonyle de la cétone qui est arraché par l'ion HO-
(voir le mécanisme de l'aldolisation)
et c'est donc la fonction aldéhyde qui conduit à la fonction alcool dans le cétol formé:
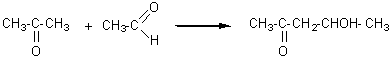 4-hydroxypentan-2-one 4-hydroxypentan-2-one
Cette réaction a lieu
en milieu basique .
|
|
CHICHIBABIN
C'est une substitution
nucléophile sur le cycle pyridinique par suite d'un
traitement par l'amidure de sodium dans l'ammoniac liquide.
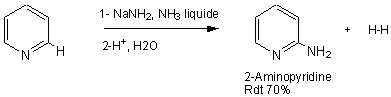
|
|
CLAISEN(Condensation)
Les esters dont l'atome de carbone en α
du groupement fonctionnel porte un ou des atomes d'hydrogène, peuvent subir
une réaction d'autocondensation qui conduit à un β-céto-ester
:
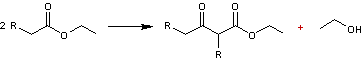
La réaction a
lieu en milieu basique fort: éthanolate
de sodium
Mécanisme:

On peut aussi faire réagir une cétone et un ester en milieu fortement basique
(C2H5O-) et on aboutit par le même mécanisme
à une β-dicétone.
|
|
CLAISEN(Réarrangement)
Dans un solvant peu polaire,
l'action d'un ion phénolate sur un halogénure d'allyle qui devrait conduire à
un éther oxyde allylique conduit en fait à un phénol substitué, par suite
d'un réarrangement appelé réarrangement de Claisen:
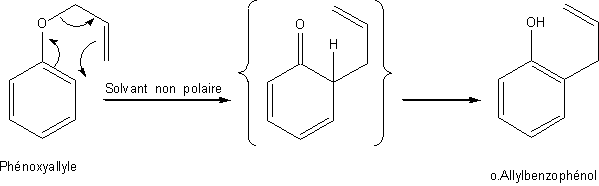
|
|
REDUCTION DE CLEMMENSEN
Réduction d'un
aldéhyde ou d'une cétone.
Un aldéhyde et une cétone
peuvent être réduites totalement, le groupement carbonyle CO devenant CH2. On utilise comme
réducteur, un amalgame de zinc et de l'acide chlorhydrique concentré.Cette méthode n'est
pas applicable dans les cas ou le composé carbonylé
contient des groupes sensibles aux acides. On utilise alors la méthode de Wolff-Kishner.
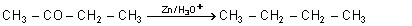
Cette méthode est appliquée
notamment lorsqu'on souhaite greffer une chaîne saturée linéaire de plus de 3
carbones, sur un noyau aromatique.La
réaction de Friedel et Crafts
menée avec l'halogénoalcane correspondant à la
chaîne linéaire, conduit à un mélange dû à l'isomérisation du carbocation
intermédiaire. On procède à une acylation
(fixation de R-CO- grâce à R-CO-Cl) puis à une réduction de Clemmensen de la cétone obtenue.
|
|
REARRANGEMENT DE
CORNFORTH
|
|
REARRANGEMENT DE COPE
Ce réarrangement se produit avec les molécules comportant un diène 1-5.
Il s’agit d’un réarrangement qui s’apparente à celui de Claisen pour les composés
non oxygénés.
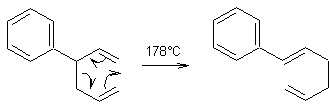
Arthur C.Cope
(1909-1966) a été Professeur au M.I.T.
|
|
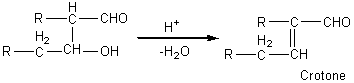
CROTONISATION
Déshydratationd'un aldol ou d'un cétol .
Un aldol ou un cétol, formé lors d'une aldolisation ou d'une cétolisation, placé en milieu acide et à température
ordinaire (ou plus élevée) se déshydrate en donnant une crotone
(aldéhyde ou cétone ayant une double liaison conjuguée avec la double liaison
du carbonyle) :
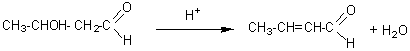
|
|
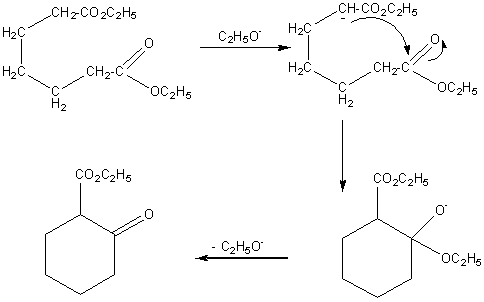
DIECKMANN(Condensation de)
C'est une condensation
comparable à celle de Claisen
qui intervient avec les diesters, condensation
intramoléculaire qui a lieu si le cycle formé est sans "contrainte"
c'est-à-dire si c'est un cycle à 5 ou 6 sommets(de
type cyclopentane ou cyclohexane). Avec l'heptanedioate
de diéthyle par exemple on obtient le
2-oxocyclohexanecarboxylate d'éthyle.
|
|
DIELS-ALDER
Une des réactions les
plus utiles en synthèse organique est l'addition 1-4 d'un alcène sur un diène
conjugué; elle est appelée réaction de Diels-Alder.
Exemple:
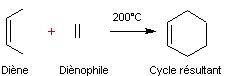
L'exemple ci-dessus
est intéressant par sa simplicité (il est pédagogique), mais le rendement de
cette réaction est très faible (environ 20%). Il faut dans une telle synthèse
utiliser un diène riche en électrons (par exemple substitué par des groupes
électrodonneurs) et un diénophile pauvre en
électrons (par exemple un alcène substitué par des groupes électroattracteurs:
voir tableau ci-dessous).
Des diènophiles très utilisés dans les
réactions de Diels-Alder
|
Tétracyanoéthylène
|
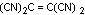
|
Acrylonitrile
|
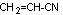
|
|
Crotonaldéhyde
|
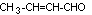
|
Acroléine
|
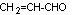
|
|
Acide cinnamique
|
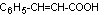
|
Acrylate d'éthyle
|
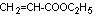
|
|
Anhydride maléique
|
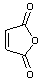
|
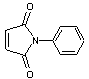
N-phénylmaléimide
|
|
|
Maléate de diméthyle
|
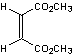
|
Fumarate de
diméthyle
|
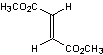
|
Des diènes souvent utilisés dans les réactions de Diels-Alder
|
Buta-1,3-diène
|
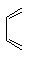
|
2,3-diméthylbuta-1,3-diène
|
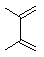
|
|
Cyclopenta-1,3-diène
|
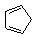
|
Cyclohexa-1,3-diène
|
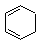
|
|
Exemple
|
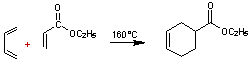
|
Le diénophile utilisé dans cette
réaction est l'acrylate d'éthyle. Le temps de réaction nécessaire à un bon
rendement (>90%) est élevé (> 12h).
En terme de mécanisme, la réaction de Diels Alder s'effectue en une seule étape, c'est une réaction
concertée.
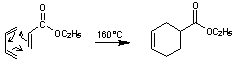
La réaction de Diels Alder est
stéréospécifique :

L' addition du maléate de diméthyle qui a
des groupes esters cis (ou Z), sur le 1,3-butadiène, on obtient un
cyclohexène cis (ou Z) substitué. Il y a rétention de la configuration du diénophile. La stéréochimie du diènophile
est conservée et avec un autre diène on montrerait que celle du diène l'est
aussi.
Une cycloaddition (réaction de Diels-Alder
avec un diène cyclique par exemple) conduit au dérivé endo seulement.
|
|
REACTION DE DOEBNER-KNOEVENAGEL
Cette réaction connue
aussi sous le nom de réaction de Knoevenagel
permet de passer d'un aldéhyde à un acide a-éthylénique,
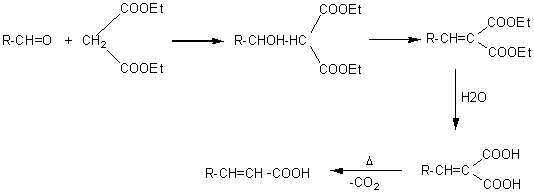
Mais aussi:
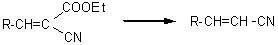
et
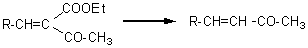
|
|
REACTION DE DOEBNER
ET MILLER
C'est une réaction
permettant la synthèse des dérivés de la quinoléïne
selon un procédé identique à celui utilisé par Skraup mais en
condensant des crotones, c'est à dire des aldols
déshydratés, avec l'aniline:
Exemple:
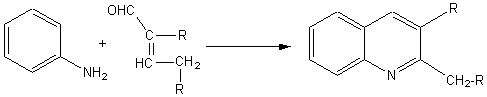
Si R = H on obtient l' a méthylquinoléïne (quinaldine).
|
|
REACTION DE FRIEDEL
ET CRAFTS
Il s'agit de
l'alkylation ou de l'acylation d'un noyau aromatique (Benzène
ou arènes)
Alkylation :
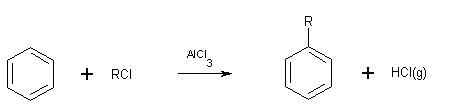
Mécanisme:
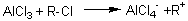
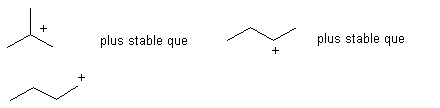

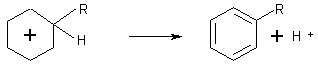
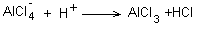
Acylation:
L'agent
électrophile est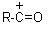
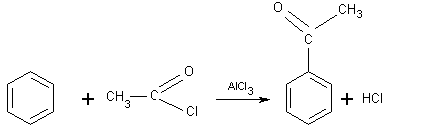
|
|
REACTION DE FRIEDLÄNDER
Elle conduit à la synthèse des quinoléines.
On fait réagir une amine aromatique ayant un carbonyle en ortho avec un
autre composé carbonylé ayant au moins deux hydrogènes en α.
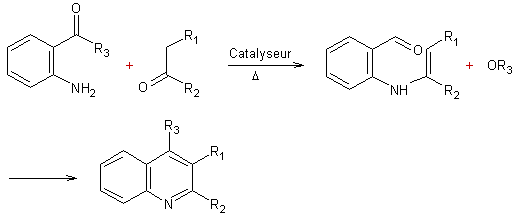
Le catalyseur peut être l'acide trifluoracétique,
l'acide toluènesulfonique, l'iode ou les acides de
Lewis.
|
|
REARRANGEMENT DE FRIES
Réarrangement d'un
ester phénolique en une hydroxyarylcétone catalysé
par un acide de Lewis.
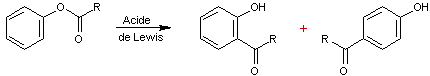
Mécanisme :
Pour le dérivé ortho :
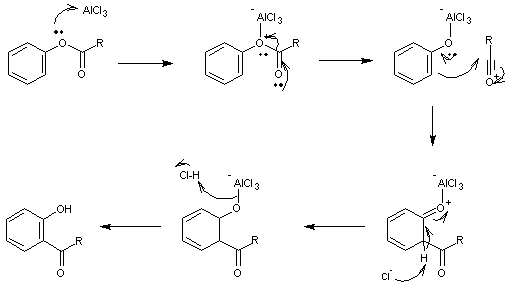
Exemple de réaction :
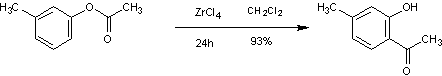
|
|
SYNTHESE DE GABRIEL,
DES AMINES
Une synthèse des
amines primaires sans mélange.
Les méthodes d'Hofmann et de Sabatier de synthèse des amines conduisent à des mélanges.
Pour éviter cela , Gabriel a mis au point une méthode, permettant de
préparer des amines primaires, seules.
Cette synthèse tient
en 3 étapes:
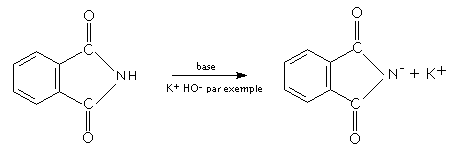
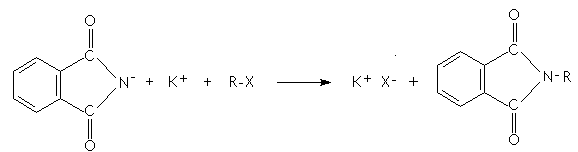
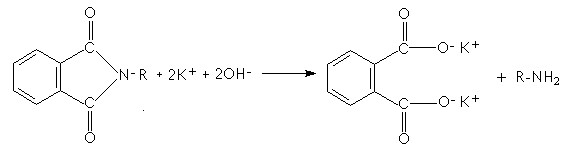
La molécule clé de
cette synthèse est le phtalimide (amide secondaire
de l'acide orthophtalique) obtenu par action de
l'ammoniac sur l'anhydride orthophtalique:
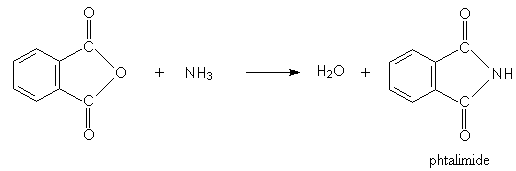
elle permet de bloquer
deux des trois hydrogènes de l'ammoniac.
|
|
GATTERMANN-KOCH
Synthèse d'aldéhydes
aromatiques.
A température
ordinaire, le monoxyde de carbone, réagissant en présence de chlorure
d'hydrogène, sur du benzène, avec AlCl3 comme catalyseur,
conduit au benzaldéhyde. Cette réaction est une oxosynthèse.
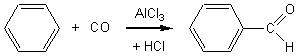
Il y a formation du
chlorure d'acyle HCOCl par action de HCl sur le monoxyde de carbone, puis une réaction de
Friedel et Crafts du chlorure d'acyle formé sur le
benzène en présence d'AlCl3 comme catalyseur.
|
|
HAJOS-PARRISH
Aldolisation énantiosélective intramoléculaire
Exemple
Une tricétone achirale est soumise à un catalyseur chiral (la
S-(-)-Proline) le solvant étant la N,N-diméthylformamide (DMF). On
obtient la dicétone chirale dite de Hajos-Parrish
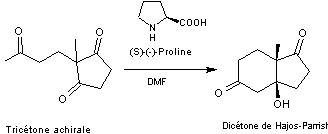
|
|
HALLER-BAUER
Alkylation de Haller-Bauer
Cette alkylation n’est utilisée en synthèse que pour les préparations
de cétones fortement substituées comme l’hexaméthylpropane,
difficiles à obtenir autrement :
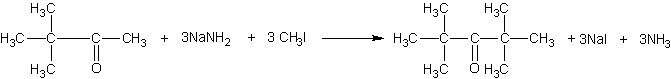
|
|
HANTZSCH(Synthèse pyridinique)
C'est la méthode
usuelle de préparation des dérivés de la pyridine:
Condensation de
l'oxo-3-butanoate d'éthyle (acétylacétate d'éthyle)
avec un aldéhyde, puis condensation du composé obtenu avec l'ammoniac
conduisant à une dihydropyridine. Après oxydation de cette molécule suivie de
l'hydrolyse de la fonction ester et d'une décarboxylation on obtient une alkyldiméthylpyridine.
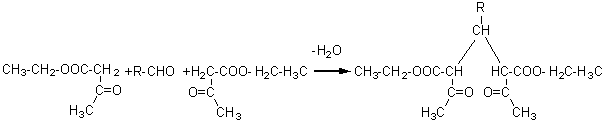
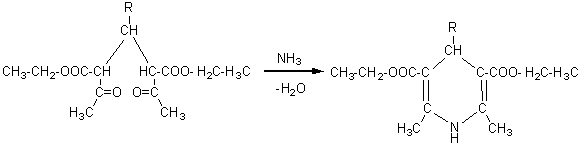
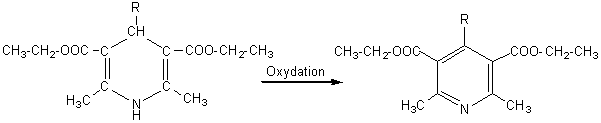
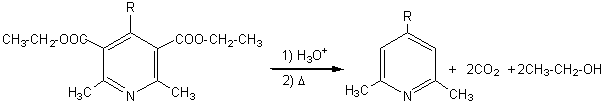
|
|
HANTZSCH (Synthèse du pyrrole)
Un β-céto ester réagit sur une α-chlorocétone ou un α-chloro-aldéhyde
en présence d'ammoniac ; on obtient un dérivé du pyrrole :
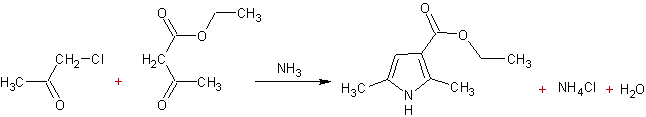
|
|
HECK (Réaction de)
La réaction dite de Heck peut se résumer par :
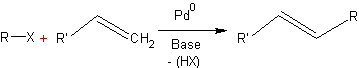
Ces réactions à base de Palladium catalyseur, mises au point par Heck, Negishi et Suzuki ont permis la synthèse de composés
importants ; elles ont permis par exemple la synthèse totale de la diazonamide A substance permettant l'inhibition de
l'assemblage de la tubuline et utilisée notamment contre les tumeurs du
côlon. La diazonamide A a
été découverte dans une ascidie (Diazona angulata).
Le palladium joue bien le rôle de catalyseur ainsi que le montre le
cycle suivant résumant la réaction
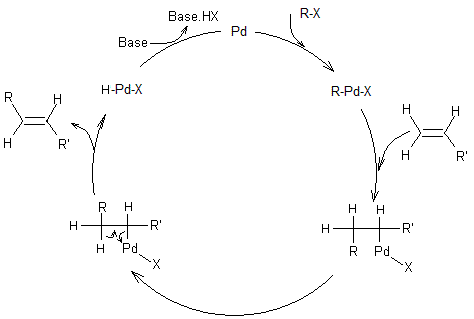
|
|
HELL-VOLHARD-ZELINSKY
Bromation d'un carbone adjacent au groupe carboxyle
d'un acide.
Exemple:

PBr3nécessaire est fabriqué in situ par action du dibrome sur des traces de phosphore rouge.
Mécanisme:

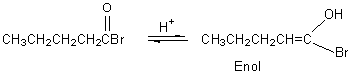
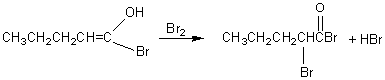
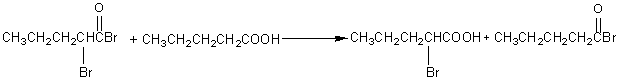
|
|
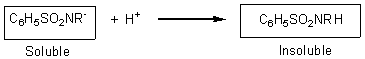
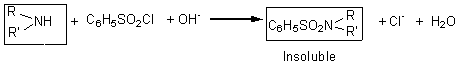
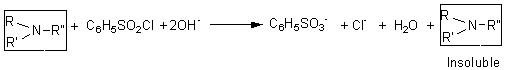
HINSBERG (test)
Permet de différencier
les amines primaires secondaires ou tertiaires : il est basé sur la
réactivité de ces amines à l'égard du chlorure de benzènesulfonyle
en milieu basique.
Les amines primaires
forment un sel de sodium soluble dans l'eau.
Les amines secondaires
forment des sulfonamides insolubles dans l'eau.
Avec les amines
tertiaires tout se passe comme s'il n'y avait pas de réaction.
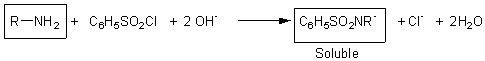
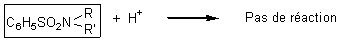
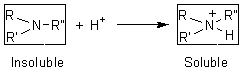
|
|
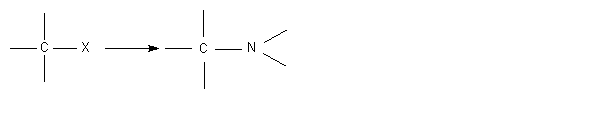
HOFMANN
Il s'agit d'une
réaction permettant la formation d' amines à partir
d'un halogénoalcane:
Réactions:
Il s'agit de
substitutions nucléophiles, le réactif nucléophile étant l'azote de l'ammoniac
ou de l'amine réagissant sur l'halogénoalcane.

mais la réaction ne s'arrête pas là et l'amine
primaire formée peut réagir (comme NH3) avec
une nouvelle molécule d'halogénoalcane R-X pour
donner une amine secondaire R-NH-R qui peut elle
même donner naissance à l'amine tertiaire R3N. On
obtient donc un mélange d'amines et non l'amine primaire seule :
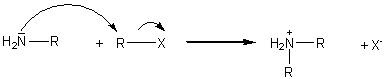
|
|
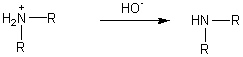
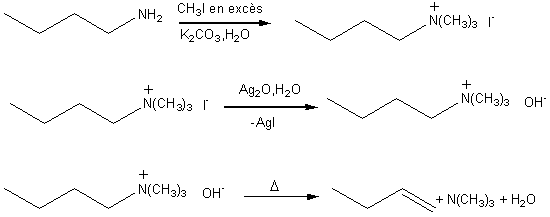
HOFMANN
L'élimination d' Hofmann permet de passer d'une amine transformée en
ammonium quaternaire (à l'aide d'un halogénoalcane,
généralement l'iodure de méthyle, (méthylation exhaustive)) que l'on traite
par une base forte (Ag2O en présence d'eau) pour obtenir un hydroxyde
d'ammonium quaternaire, qui se dégrade sous l'action de la chaleur en alcène.
Elle a été utilisée pour élucider les structures de produits naturels azotés
tels que les alcaloïdes.
|
|
HUNSDIECKER
Permet de transformer
un acide carboxylique en un bromoalcane ayant un
atome de carbone en moins.

Exemple : L'acide décanoïque
CH3-(CH2)8-COOH est transformé en 1-bromononane CH3-(CH2)8-Br
Cette réaction porte
le nom de Hunsdiecker, mais a été mise au point par
Aleksandr Borodine.
|
|
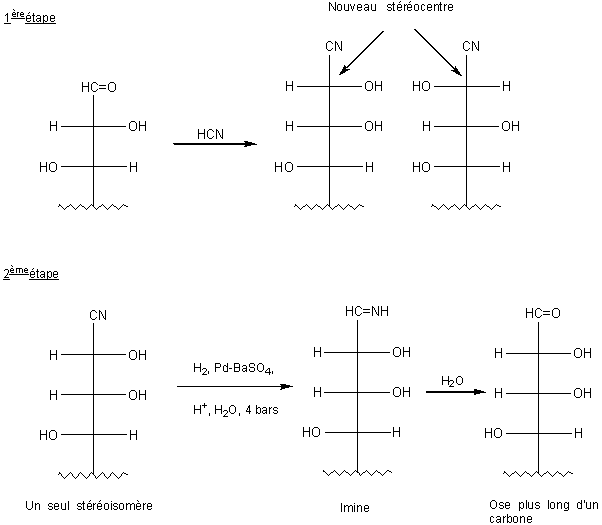
KILIANI-FISCHER
Il s'agit d'une
réaction permettant d'allonger la chaîne d'un aldose, d'un carbone.
2 étapes:
- Formation d'une
cyanhydrine par action de HCN sur la fonction aldéhyde, ce qui crée un
nouveau stéréocentre. On trie les diastéréoisomères.
- Réduction puis
hydrolyse pour retrouver une fonction aldéhyde, avec au passage formation
d'une imine.
|
|
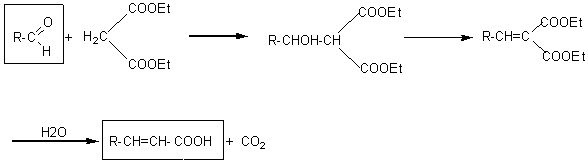
KNOEVENAGEL
Par une réaction
appelée aussi réaction de Doebner-Knoevenagel
qui s'apparente à une synthèse malonique on passe
d'un aldéhyde à un acide a-éthylénique avec allongement de la chaîne carbonée de deux unités:
On peut aussi, en
faisant réagir l'aldéhyde sur l'ester nitrile malonique préparer un nitrile
a-éthylénique.
Et par réaction de
l'aldéhyde sur un céto-ester malonique
obtenir une cétone a-éthylénique:
|
|
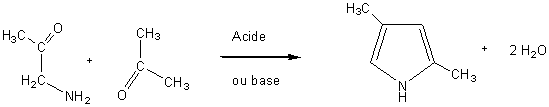
SYNTHESE DE KNORR
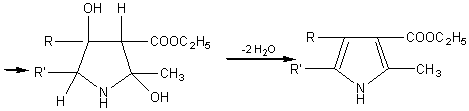
Permet de péparer les dérivés pyrroliques par action des a-amino-cétones
sur les esters b-cétoniques (ou les cétones):
Exemple:obtention du 2,4-diméthylpyrrole
Exemple:
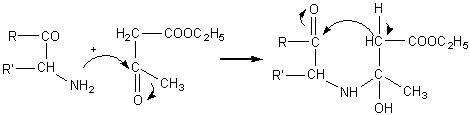
|
|
KOLBE
Il s'agit d'une
réaction de carboxylation du benzophénol (son sel
de sodium) sous l'action de CO2 à 125°C et sous pression (5bars):
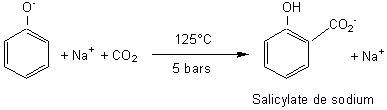
Remarque:
A température
ordinaire le benzophénate de sodium absorbe le
dioxyde de carbone pour donner le phénylcarbonate
de sodium:
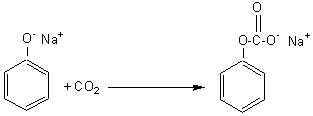
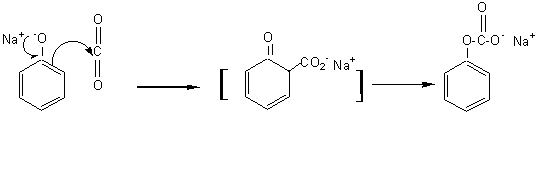
Si l'on chauffe il se produit
un réarrangement qui conduit au salicylate de sodium.
Le salicylate de
sodium est la matière première pour la synthèse de l'aspirine.
Mécanisme:
|
|
SYNTHESE MALONIQUE
Il s'agit de la
synthèse d'un acide à partir d'un dérivé halogéné, avec augmentation du
nombre de carbones, qui fait intervenir le malonate d'éthyle (propanedioate de diéthyle).
Le schéma réactionnel
est le suivant :
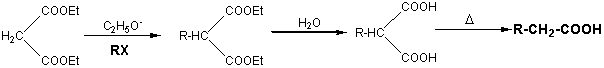
Les équations
chimiques correspondantes:
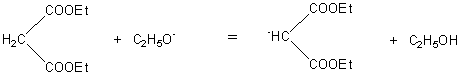
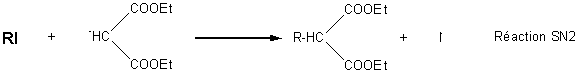

Souvent
malheureusement il y a une dialkylation et l'on
aboutit à un mélange de deux produits difficiles à séparer ensuite. Cette dialkylation n'a pas lieu facilement si le groupement R
est volumineux.
|
|
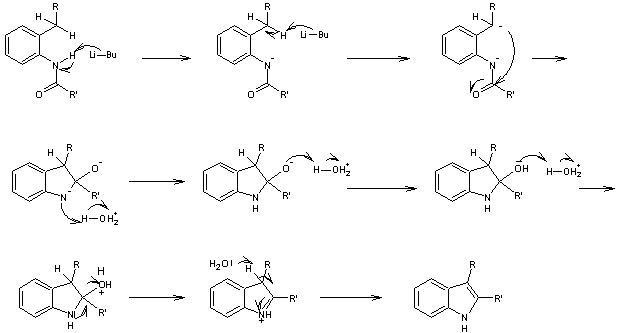
REACTION DE MADELUNG (1912) (Madelung
Walter Chimiste allemand né en 1879 et mort en 1963)
Il s'agit d'une
cyclisation intramoléculaire d'un N-phénylamide
aboutissant à l'indole ou à un de ses dérivés.
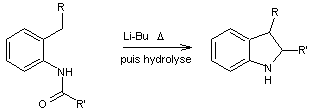
Le mécanisme de cette
réaction est le suivant :
|
|
MANNICH
Greffe d'un groupe dialcoylaminométhyle 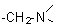 sur différentes familles de molécules (alcynes, phénols,
cétones..) en utilisant comme réactif une amine non tertiaire sur différentes familles de molécules (alcynes, phénols,
cétones..) en utilisant comme réactif une amine non tertiaire 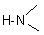 en présence de formol
(méthanal en solution): en présence de formol
(méthanal en solution):
Exemple 1: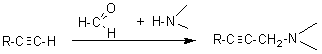
Exemple 2: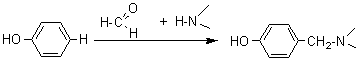
Exemple 3: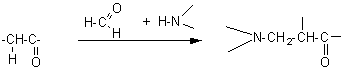
Exemple 4: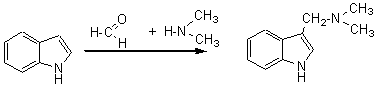
Indole
Cette dernière
réaction de synthèse conduit à la gramine
(alcaloïde toxique que l'on trouve dans de nombreuses plantes (La canne de provence ou Arundo donax, la
baldingère faux-roseau ou alpiste faux-roseau Phalaris arundinacea...))
qui a été isolée pour la première fois des feuilles d'orge (Hordeum vulgare)
par Hans Von Euler et Erdtmann.
Le mécanisme de cette
réaction est le suivant :
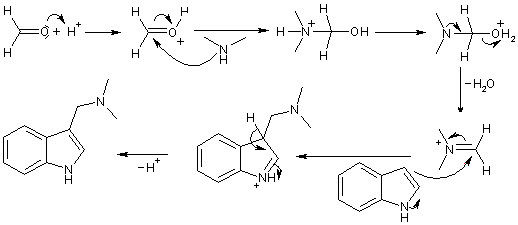
|
|
MICHAEL
Addition (ou réaction) de Michael
La réaction de Michael est une addition
nucléophile d’un composé nucléophile (souvent un énolate ou une amine)
appelé donneur de Michael sur une double liaison activée par un
groupement électroattracteur, comme un groupe carbonyle appelé accepteur de
Michael.
Elle permet la formation de liaisons C–C (ou
C–N) en β-position d’un groupe carbonyle.
Type de réaction : Addition 1,4 .
Double liaison activée typique : CH₂=CH–CO–R, CH₂=CH–CN, etc.
Exemple
1
Penta-2,4-dione sur acroléine
Réactifs :
- Nucléophile : CH₃–CO–CH₂–CO–CH₃ (penta-2,4-dione)
- Électrophile : CH₂=CH–CHO (acroléine)
Étapes :
- Le penta-2,4-dione forme
un énolate sur le carbone central (entre les deux carbonyles).
- Cet énolate attaque
l'acroléine sur sa double liaison activée.
Résultat : formation d’un composé 1,5-dicarbonylé avec une
fonction aldéhyde en bout.
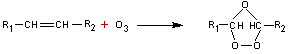
Exemple
2
Acrylate de méthyle sur éthane diamine
Réactifs :
- Nucléophile : NH₂–CH₂–CH₂–NH₂ (éthanediamine)
- Électrophile : CH₂=CH–COOH
(acide acrylique)
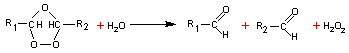
Le groupe amine restant libre peut à son tour réagir sur une
molécule d’acide acrylique pour donner une éthane diamine disubstituée.
|
|
OZONOLYSE
L'ozonolyse d'un
alcène consiste à le traiter par l'ozone pour
former un ozonide qui est ensuite réduit par du zinc dans l'acide acétique ou
par réaction avec le sulfure de diméthyle.
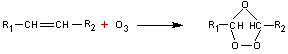
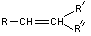
On aboutit à des composés carbonylés :
- Si l'alcène de
départ est du type
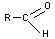
On obtient un aldéhyde
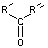
et une cétone
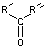
Si l'alcène de départ
est du type
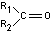
on obtient deux cétones
|
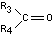
|
et
|
|
La nature des composés
carbonylés obtenus permet d'identifier l'alcène de départ.
En l'absence de zinc,
l'oxydation de ou des aldéhydes par l'eau oxygénée conduit à un ou des
acides.
|
|
SYNTHESE DE PAAL-KNORR
Permet la préparation
des systèmes hétérocycliques à 5 sommets.
Elle consiste à chauffer un composé 1,4-dicarbonylé énolisable avec, soit un agent déshydratant (acide
sulfurique par exemple, ou P2O5), soit l'ammoniac ou une amine, soit un sulfure (P2S5), pour obtenir soit un furanne, soit un
pyrrole, soit un thiophène:
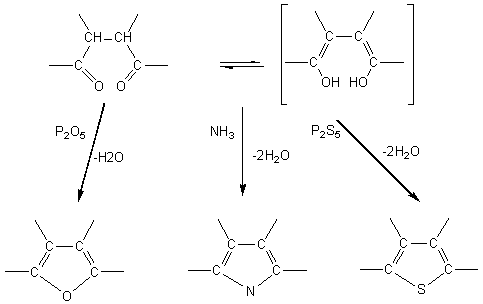
Le composé 1,4-dicarbonylé pouvant être une dicétone, un diacide, ou un acide cétonique
Exemple: synthèse du N-1-méthyléthyl-2,5- diméthylpyrrole
à partir de l' hexan-2,5-dione et de la
propan-2-amine.

Exemple :synthèse du
2,5-diphénylthiofène à partir du 1,2-dibenzoyléthane.
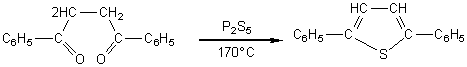
|
|
REACTION DE PAULY
Elle permet de
détecter deux acides aminés car ils donnent une coloration rouge avec le sel
de diazonium de l'acide sulfanilique.
Ce sont
- La tyrosine
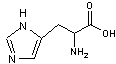
- L'histidine
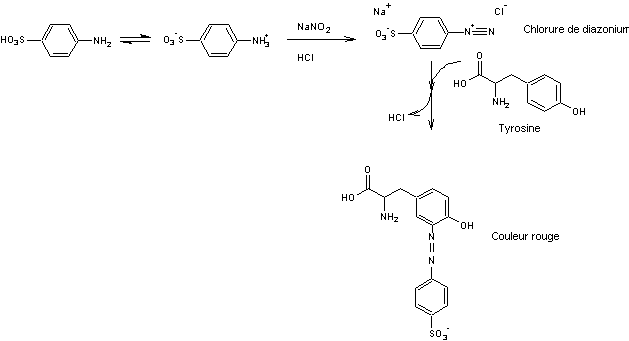
Equations de réaction
avec la tyrosine
|
|
REACTION DE PERKIN
Condensation du benzaldéhyde
avec l'anhydride éthanoïque en présence d'éthanoate de sodium, pour donner de
l'acide cinnamique (Cet acide se trouve par ailleurs dans certaines plantes
comme Styrax benzoe (originaire
d'Indonésie).
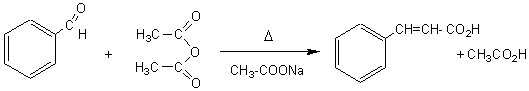 . .
Il s'agit d'une
addition nucléophile de -CH2-COOH sur le
carbonyle.
Des condensations
semblables se produisent entre le benzaldéhyde et des aldéhydes saturés (mécanisme de l'aldolisation;
crotonisation):
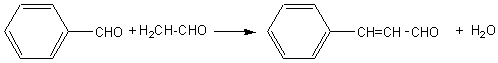
conduisant ici à
l'aldéhyde cinnamique;
ou entre le
benzaldéhyde et les cétones (mécanisme de la cétolisation; crotonisation):
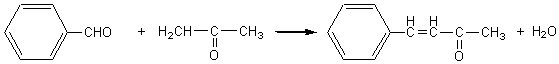
conduisant ici à la benzylidène acétone.
Remarque: l'acide coumarique est l' acide o.hydroxycinnamique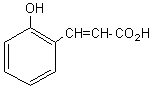 dont la lactone
(ester cyclique) est la coumarine dont la lactone
(ester cyclique) est la coumarine 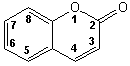 . Cette dernière molécule a été préparée pour la première fois
en 1868 par W.H.PERKIN senior. . Cette dernière molécule a été préparée pour la première fois
en 1868 par W.H.PERKIN senior.
L'acide coumarique est un indicateur de pH, fluorescent (incolore
à pH 7,2 il devient fluorescent vert à pH 9,0).
L'acide p.hydroxycinnamique (ou
4-hydroxycinnamique) existe également :

|
|
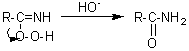
RADZISZEWSKI
L'hydratation des
nitriles conduit aux amides, mais il n'est pas facile de s'arrêter à ce stade
car l'amide formé s'hydrolyse assez facilement en
acide.
En traitant le nitrile
par le peroxyde d'hydrogène (eau oxygénée) il y a formation d'un hydroperoxyde qui se décompose en amide en milieu
alcalin.
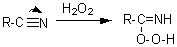
|
|
REFORMATSKY
Il s'agit de la
réaction d'un composé organique halogéné, généralement un a-bromoester,
avec un aldéhyde ou une cétone en présence de zinc métal. Il y a d'abord
formation d'un intermédiaire organozincique de
l'ester, qui s'additionne ensuite au groupe carbonyle du composé carbonylé.
Le résultat est un b-hydroxyester qui
peut en général être déshydraté pour donner un ester insaturé.
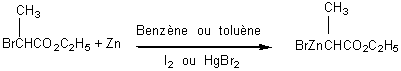
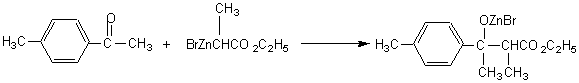
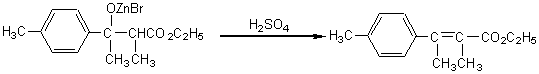
Il
est parfois nécessaire d'activer le zinc en ajoutant un petit cristal d'iode
de bromure de mercure(II) ou du cuivre. L'avantage
du zinc sur le magnésium, est que l'organomagnésien de l' a-bromoester tend à attaquer le groupe CO d'une autre
molécule d'ester ce qui n'a pas lieu avec l'organozincique.
|
|
REIMER ET TIEMANN
Formylation d'un phénol par action du trichlorométhane
(chloroforme) en milieu basique
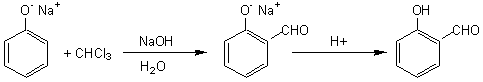
Ou

dont
le mécanisme est :

|
|
REPPE
Addition de monoxyde
de carbone (CO) en présence d'eau sur la double liaison d'un alcène pour
conduire à un acide à chaîne plus longue.
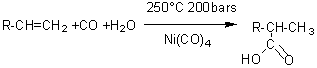
Le nickel carbonyle
joue le rôle de catalyseur.
Cette réaction porte
le nom d'oxosynthèse.
|
|
RILEY
On peut transformer la
butanone en butanedione,
par oxydation avec le dioxyde de sélénium, c'est la réaction de Riley :

|
|
RITTER
Obtention d'amides
primaires N-substitués.
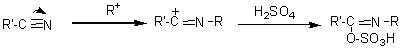
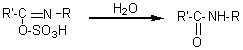
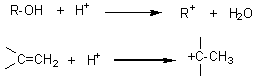
Remarque: Avec K-CN on obtient
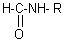
Le carbocation R+
est obtenu à partir d'un alcool R-OH ou à partir d'un alcène par action
de l'acide sulfurique concentré ou en présence de AlCl3 :
|
|
ROBINSON (Annellation de)
Il s'agit d'une addition de Michael suivie d'une condensation aldolique intramoléculaire:
Exemple:
Voir la synthèse de l'isophorone.
Voir aussi l' exercice 93
|
|
ROSENMUND
Les chlorures d'acyles
hydrogénés par le dihydrogène H2 en présence de
palladium désactivé, conduisent à des aldéhydes.
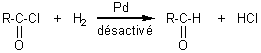
|
|
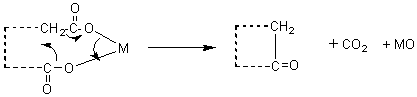
RUZICKA
Permet la préparation
des grands cycles ou macrocycles.
On fait appel aux
diacides ou à leurs dérivés, diesters ou
dinitriles.
La pyrolyse des
diacides ou de leurs sels de métaux lourds (M = Pb,Th,Ce...) conduit à des cyclanones
(cétones cycliques) avec des rendements faibles (environ 5%). On peut ensuite
réduire ces cétones pour passer aux hydrocarbures.
|
|
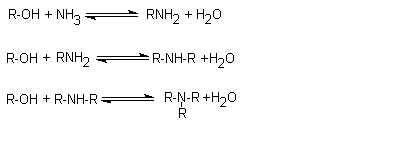
SABATIER
C'est l'action d'un
alcool sur l'ammoniac vers 300°C en présence de thorine (ThO2). Comme avec la méthode d'Hofmann on
obtient un mélange d'amines
|
|
SABATIER ET MAILHE
Il s'agit d'une
réduction des acides carboxyliques en aldéhydes en utilisant l'acide
méthanoïque (acide formique) comme réducteur.
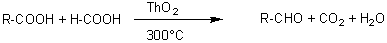
Il se forme en même
temps la cétone R-CO-R
|
|
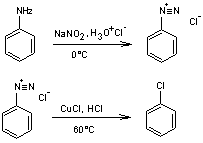
SANDMEYER
Cette réaction permet d'obtenir
des chloro ou bromo
arènes en traitant un sel de diazonium de ces
arènes par l'acide chlorhydrique ou bromhydrique, en présence d'un catalyseur
qui est le chlorure de cuivre(I) ou le bromure de cuivre(I).
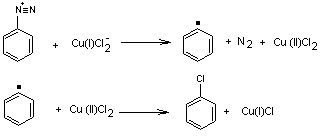
Mécanisme :
Le mécanisme de cette réaction est complexe et fait intervenir
des radicaux :
Remarque: L'obtention des dérivés iodés peut se faire sans catalyseur, en
utilisant l'iodure d'hydrogène, tandis que les dérivés fluorés se font par
voie indirecte (réaction de Schiemann).
|
|
SCHIEMANN
Permet la préparations des fluoroarènes,
par voie indirecte par une réaction un peu semblable à celle de Sandmeyer, mais sans catalyseur. On passe par un tétrafluoroborate de diazonium:
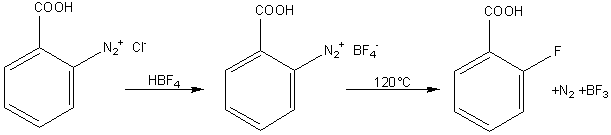
La sytnhèse
directe du dérivé fluoré est impossible à cause du caractère très
exothermique de la réaction.
|
|
SENDERENS
Préparation de cétones
à partir d'acides carboxyliques.
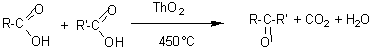
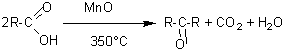
|
|
SKRAUP
Réaction de
cyclisation aboutissant à la quinoléïne ou à ses dérivés.
Il s'agit d'une
réaction de condensation d'un aldéhyde ou d'une cétone a,b-insaturé avec
l'aniline ou une amine aromatique primaire ayant une position ortho libre en
milieu acide et en présence d'un agent oxydant.
Exemple 1:
L'acroléïne
est l'aldéhyde a,b-insaturé et l'aniline est l'amine aromatique.
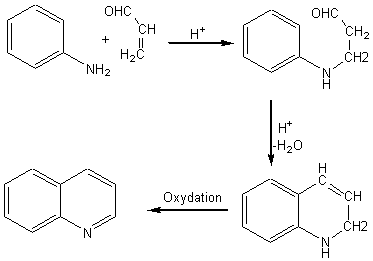
Le
composé obtenu est la quinoléïne.
Skraup avait effectué cette synthèse en 1879 en
chauffant du glycérol avec de l'aniline en présence d'acide sulfurique. En
fait le glycérol se déshydrate en
acroléïne dans un premier temps.
Exemple
2:
La méthylvinylcétone est la cétone a,b-insaturée
et l'aniline est l'amine aromatique.
Le
composé obtenu est la g-méthylquinoléïne ou lépidine.
Les
acides qui sont utilisés sont les acides sulfurique ou phosphorique;
les oxydants couramment employés sont les dérivés nitrés correspondant aux
amines employées (ainsi avec l'aniline, l'oxydant est le nitrobenzène); ainsi après avoir joué le rôle d'oxydant le dérivé
nitré est réduit en l'amine utilisée dans la condensation.
|
|
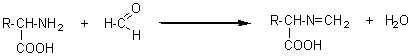
SÖRENSEN(méthode de)
Méthode de dosage d'un
acide aminé: on transforme la fonction amine en
imine par action du méthanal en solution (formol) et la fonction carboxyle
devient alors dosable par la soude en présence de phénolphtaléine.
|
|
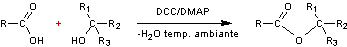
STEGLICH (Estérification de )

On utilise le dichlorométhane (CH2Cl2)
comme solvant, le dicyclohexylcarbodiimide (DCC) comme agent de
couplage et le diméthylamidopyridine (DMAP) comme
catalyseur.
C'est une réaction
douce qui convient aux alcools stériquement
encombrés comme par exemple les tertiobutanols
(esters tertiobutyliques) qui ont tendance dans les
conditions habituelles d'estérification à former des carbocations et de
l'isobutène.
Mécanisme :
|
|
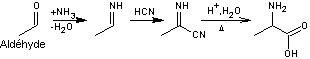
STRECKER
- La synthèse de Strecker permet d'obtenir des acides aminés à partir
d'aldéhydes.
Exemple : Synthèse de l'alanine à partir de l'éthanal:
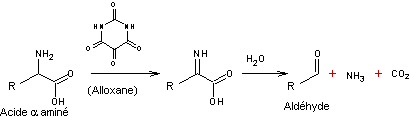
- La dégradation de Strecker permet d'obtenir un aldéhyde à partir
d'acides aminés.
Exemple général :
|
|
COUPLAGE DE SUZUKI
Le principe est le suivant :
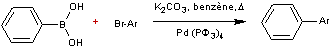
En pratique :
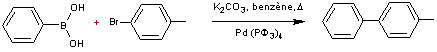
Exemple :
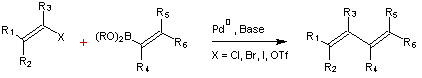
Remarque :
Ce type de couplage mis au point par Suzuki (Prix Nobel en 2010) a
vu son champ d'action considérablement élargi. On peut coupler aussi des
alkyles, des alcényles, des alcynyles. A la place
de l'acide boronique, on peut utiliser des esters boroniques, mais aussi des organoborates.
Exemple : Couplage de Suzuki-Miyaura
|
|
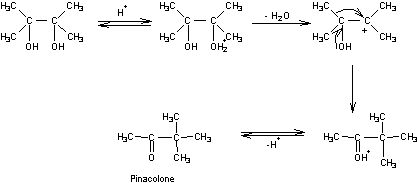
TRANSPOSITION
PINACOLIQUE
Transposition se produisant lors de la déshydratation du pinacol (un glycol) et qui conduit à une cétone, la pinacolone :
|
|
VAN SLYKE
La méthode de Van Slyke de dosage des acides a-aminés consiste à
traiter la molécule avec de l'acide nitreux, HNO2 :
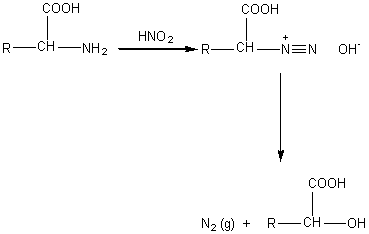
Du volume de diazote
dégagé on en déduit la quantité d'acide aminé au départ.
|
|
WILGERODT
La réaction de Wilgerodt est un réarrangement (suivi d'une oxydation)
que subissent les cétones aromatiques a (la fonction cétone est en a du noyau aromatique)en présence de polysulfure d'ammonium (NH4S2) ou de soufre et d'une amine secondaire
(souvent la morpholine). Il se forme un w-acide:
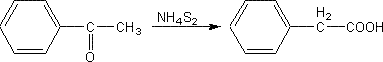

|
|
WILLIAMSON
La synthèse de
Williamson permet d'obtenir des éther-oxydes par action d'un alcoolate ou
d'un phénolate, sur un dérivé halogéné.
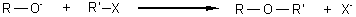
|
|
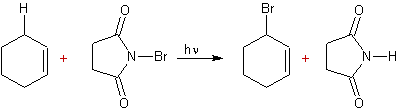
WOHL-ZIEGLER
Il s'agit de la bromation d'un alcène par le dibrome en présence de N-bromosuccinimide
; c'est une réaction en chaîne de type radicalaire.

Mécanisme :
L'initiation se fait par l'intermédiaire de l'azo-bis-isobutyronitrile qui chauffé donne un radical.

|
|
WOLFF-KISHNER
Il s'agit de la
désoxygénation de la fonction carbonyle : C=O transformé en CH2.
1èreétape : on forme l'hydrazone du composé carbonylé
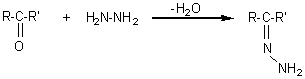
2èmeétape : On chauffe l'hydrazone à température élevée en présence d'une base,
elle se décompose avec dégagement d'azote:
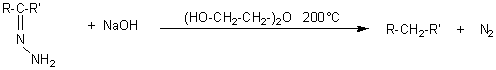
Remarque 1: (HO-CH2-CH2-)2-O corespond
au diéthylène glycol.
Remarque 2: Cette opération
s'adresse aux composés carbonylés stables en milieu basique. Pour ceux qui
sont stables en milieu acide, il faut opérer selon la technique de Clemmensen.
|
|
WURTZ
Le sodium en agissant
sur un iodure d'alkyle fournit un alcane.Il
s'agit d'une méthode permettant d'allonger une chaîne carbonée.
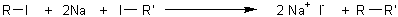
|
|
ZEREWITINOFF
Un organomagnésien R-MgX est décomposé par des réactifs à hydrogène mobile A- H+ selon
l'équation

Si on utilise CH3MgI on aura un dégagement de CH4 et de la mesure du volume de méthane recueilli on pourra en
déduire la quantité de matière de AH initialement présente. Il s'agit d'un
dosage de AH.
Les différents A-H à
hydrogène mobile sont regroupés dans le tableau:
|
Eau
|
OH-H
|
|
Alcools
|
RO-H
|
|
Phénols
|
ArO-H
|
|
Thiols
|
RS-H
|
|
Amines
|
R(R')N-H
|
|
Amides
|
RCO-N(R')-H
|
|
Acides
|
RCOO-H
|
|